SECRETARIA DE MEDIO AMBIENTE Y RECURSOS NATURALES
NORMA Oficial
Mexicana NOM-098-SEMARNAT-2002, Protección ambiental-Incineración de residuos,
especificaciones
de operación y límites de emisión de contaminantes.
Al margen un sello con el Escudo Nacional, que dice:
Estados Unidos Mexicanos.- Secretaría de Medio Ambiente y Recursos
Naturales.
NORMA OFICIAL MEXICANA
NOM-098-SEMARNAT-2002, PROTECCION AMBIENTAL-INCINERACION DE RESIDUOS,
ESPECIFICACIONES DE OPERACION Y LIMITES DE EMISION DE CONTAMINANTES.
JUAN RAFAEL ELVIRA QUESADA,
Subsecretario de Fomento y Normatividad Ambiental de la Secretaría de Medio
Ambiente y Recursos Naturales y Presidente del Comité Consultivo Nacional de
Normalización de Medio Ambiente y Recursos Naturales, con fundamento en lo
dispuesto en los artículos 32 bis fracciones I, II IV y V de la Ley Orgánica de
la Administración Pública Federal; 8 fracción V del Reglamento Interior de la
Secretaría de Medio Ambiente y Recursos Naturales; 5 fracciones V, VI, 36, 37,
37 Bis, 137, 150, 151, 151 Bis de la Ley General del Equilibrio Ecológico y la
Protección al Ambiente; 7 fracción II, 62 y 63 de la Ley General para la
Prevención y Gestión Integral de los Residuos; 38 fracción II, 40 fracciones X,
XIII y XVII, 47 fracción I de la Ley Federal sobre Metrología y Normalización y
33 de su Reglamento, y
CONSIDERANDO
Que con fecha 8 de
septiembre de 2000, se publicó en el Diario
Oficial de la Federación el Proyecto de Norma Oficial Mexicana
PROY-NOM-098-ECOL-2000, Protección ambiental.- Incineración de residuos,
especificaciones de operación y límites de emisión de contaminantes, con el fin
de que los interesados, dentro del plazo establecido en la ley en la materia,
presentaran sus comentarios ante este Comité;
Que durante el citado plazo,
los interesados presentaron sus comentarios al proyecto en cuestión, los cuales
después de ser analizados en su totalidad por el Grupo de Trabajo, éste
consideró que algunos de ellos eran procedentes y como consecuencia se hicieron
modificaciones sustanciales al Proyecto de Norma Oficial Mexicana;
Que dichas modificaciones
fueron presentadas al Comité Consultivo Nacional de Normalización de Medio
Ambiente y Recursos Naturales (COMARNAT) en su sesión del 25 de noviembre de
2002, por lo que éste, con base en lo establecido por el artículo 33 del
Reglamento de la Ley Federal sobre Metrología y Normalización, consideró
procedente que una vez modificado sustancialmente el proyecto sea publicado
para consulta pública, de conformidad con el artículo 47 fracción I de la Ley
Federal sobre Metrología y Normalización, bajo la denominación de
PROY-NOM-098-ECOL-2002, Protección ambiental.- Incineración de residuos,
especificaciones de operación y límites de emisión de contaminantes.
Que el 23 de abril de 2003
se publicó en el Diario Oficial de la
Federación, el Acuerdo por el cual se reforma la nomenclatura de las normas
oficiales mexicanas expedidas por la Secretaría de Medio Ambiente y Recursos
Naturales, así como la ratificación de las mismas previa a su revisión quinquenal,
acción que eventualmente llevó a cambiar el nombre de la norma a
NOM-098-SEMARNAT-2002.
Que en cumplimiento a lo
dispuesto por el artículo 47 fracción I de la Ley Federal sobre Metrología y
Normalización con fecha 27 de junio de 2003 se publicó en el Diario Oficial de la Federación, con
carácter de proyecto la presente Norma Oficial Mexicana bajo la denominación
PROY-NOM-098-ECOL-2002, Protección ambiental-Incineración de residuos,
especificaciones de operación y límites de emisión de contaminantes, con el fin
de que los interesados, en un plazo de 60 días naturales, posteriores a la
fecha de su publicación presentaran sus comentarios al Comité Consultivo
Nacional de Normalización de Medio Ambiente y Recursos Naturales, sito en
bulevar Adolfo Ruiz Cortines número 4209, piso 5o., colonia Jardines en la
Montaña, código postal 14210, Delegación Tlalpan, México D.F., vía fax
5628-0632 y en el correo electrónico industria@semarnat.gob.mx.
Que de acuerdo a lo
establecido en el artículo 47 fracciones II y III de la Ley Federal sobre
Metrología y Normalización, los interesados presentaron sus comentarios al
proyecto de norma en cuestión, los cuales fueron analizados por el COMARNAT en
su sesión extraordinaria celebrada el 25 de febrero de 2004, realizándose las
modificaciones procedentes al proyecto.
Que habiéndose cumplido el
procedimiento establecido en la Ley Federal sobre Metrología y Normalización
para la elaboración de normas oficiales mexicanas, el Comité Consultivo
Nacional de Normalización de Medio Ambiente y Recursos Naturales, aprobó la
presente Norma Oficial Mexicana.
Por lo expuesto y fundado,
he tenido a bien expedir la siguiente:
NORMA OFICIAL MEXICANA
NOM-098-SEMARNAT-2002,PROTECCIÓN AMBIENTAL-INCINERACIÓN DE RESIDUOS,
ESPECIFICACIONES DE OPERACIÓN Y LIMITES DE EMISIÓN DE CONTAMINANTES
PREFACIO
En la elaboración de esta
Norma Oficial Mexicana participaron:
- ASOCIACIÓN MEXICANA DE LABORATORIOS ANALÍTICOS PARA EL
MEDIO AMBIENTE, A.C.
- ASOCIACIÓN NACIONAL DE LA INDUSTRIA QUÍMICA, A.C.
- CÁMARA NACIONAL DE LA INDUSTRIA FARMACÉUTICA
- PETRÓLEOS MEXICANOS
PEMEX PETROQUÍMICA
PETROQUÍMICA PAJARITOS, S.A. DE C.V.
- SECRETARIA DE ENERGÍA
SUBSECRETARIA DE HIDROCARBUROS
DIRECCIÓN DE SEGURIDAD Y PROTECCIÓN AL AMBIENTE
- SECRETARIA DE MEDIO AMBIENTE Y RECURSOS NATURALES
SUBSECRETARIA DE FOMENTO Y NORMATIVIDAD AMBIENTAL
SUBSECRETARIA DE GESTIÓN PARA LA PROTECCIÓN AMBIENTAL
INSTITUTO NACIONAL DE ECOLOGÍA
PROCURADURÍA FEDERAL DE PROTECCIÓN AL AMBIENTE
- SECRETARIA DE SALUD
DIRECCIÓN GENERAL DE SALUD AMBIENTAL
ÍNDICE
0. Introducción
1. Objetivo
2. Campo de aplicación
3. Referencias
4. Definiciones y terminología
5. Especificaciones
6. Recepción de los residuos
7. Operación de una
instalación de incineración
8. Medición en chimenea
9. Emisiones al ambiente
10. Evaluación de la
conformidad
11. Grado de concordancia con normas y
lineamientos internacionales y con las normas mexicanas tomadas como base para
su elaboración
12. Bibliografía
13. Observancia de la norma
Transitorios
Anexos
0.
Introducción
La Ley General del
Equilibrio Ecológico y la Protección al Ambiente establece que para la
formulación y conducción de la política ambiental y la expedición de normas
oficiales mexicanas se deben observar como principios, entre otros: que toda
persona tiene derecho a disfrutar de un ambiente adecuado para su desarrollo,
salud y bienestar; las autoridades y los particulares deben asumir la
responsabilidad de la protección del equilibrio ecológico; quienes realicen
obras o actividades que afecten o puedan afectar el ambiente están obligados a
prevenir, minimizar o reparar los daños que causen, así como asumir los costos
que dicha afectación implique. Asimismo, debe incentivarse a quien proteja el
ambiente y la prevención de las causas que generan desequilibrios ecológicos ya
que es el medio más eficaz para evitarlos.
A medida que la población y
las actividades productivas del país han ido creciendo, la generación de
residuos sólidos municipales, hospitalarios e industriales, se ha incrementado
de tal manera, que el impacto y el riesgo que ocasiona su manejo, tratamiento y
disposición final representan en la actualidad un verdadero problema, en
especial para aquellos residuos considerados como peligrosos.
Por lo tanto, es necesario
ampliar y diversificar la infraestructura y sistemas orientados a la
minimización, reutilización, reciclaje y tratamiento de residuos. Una
alternativa tecnológica de disposición es la incineración, la cual permite
reducir el volumen y peligrosidad de los mismos.
La incineración de residuos
provenientes de cualquier actividad, incluyendo los residuos peligrosos,
produce emisiones que provocan la contaminación del ambiente y con ello dañan a
los ecosistemas y la salud humana; lo cual demanda la adopción de acciones
preventivas tendientes a propiciar condiciones de operación adecuadas y valores
límite de emisión aceptables, en particular en lo que se refiere a las dioxinas
y furanos. Las acciones preventivas, de conformidad con la política ecológica,
requieren de un enfoque en el que se incluyan los diferentes medios receptores,
lo cual implica considerar de manera integral el control de las emisiones al
aire y el manejo de las cenizas.
Por lo anterior, al
publicarse esta Norma Oficial Mexicana se establece el primero de los distintos
compromisos que derivarán del Convenio de Estocolmo; ya que al establecer
límites máximos permisibles de emisiones a la atmósfera particulares para las
instalaciones de incineración existentes y nuevas en el país se está procurando
el cuidado de la salud de la población y del ambiente.
1.
Objetivo
Esta Norma Oficial Mexicana
establece las especificaciones de operación, así como los límites máximos
permisibles de emisión de contaminantes a la atmósfera para las instalaciones
de incineración de residuos.
2.
Campo de aplicación
Esta Norma Oficial Mexicana
es de observancia obligatoria aplicable en todo el territorio mexicano, con
excepción de los mares territoriales en donde la nación ejerza su jurisdicción,
para todas aquellas instalaciones destinadas a la incineración de residuos,
excepto de hornos crematorios, industriales y calderas que utilicen residuos
como combustible alterno.
No aplica para la
incineración de residuos (desechos) radiactivos, para los cuales se aplicarán
las disposiciones que al respecto emita la Comisión Nacional de Seguridad
Nuclear y Salvaguardias.
3.
Referencias
3.1 Norma Oficial
Mexicana NOM-001-SEMARNAT-1996, Que establece los límites máximos permisibles
de contaminantes en las descargas de aguas residuales en aguas y bienes
nacionales, publicándose en el Diario
Oficial de la Federación (D.O.F.), el 6 de enero de 1997, como
NOM-001-ECOL-1996, la cual cambió de nomenclatura por el Acuerdo emitido en el
D.O.F. el 23 de abril de 2003, quedando con el nombre que aparece al inicio de
esta cita.
3.2 Norma Oficial
Mexicana NOM-002-SEMARNAT-1996, Que establece los límites máximos permisibles
de contaminantes en las descargas de aguas residuales a los sistemas de
alcantarillado urbano o municipal, publicándose en el Diario Oficial de la Federación (D.O.F.), el 3 de junio de 1998,
como NOM-002-ECOL-1996, la cual cambió de nomenclatura por el Acuerdo emitido
en el D.O.F. el 23 de abril de 2003, quedando con el nombre que aparece al
inicio de esta cita.
3.3 Norma Oficial
Mexicana NOM-008-SCFI-1993, Sistema General de Unidades de Medida, publicada en
el Diario Oficial de la Federación
el 14 de octubre de 1993.
3.4 Norma Oficial
Mexicana NOM-052-SEMARNAT-1993, Que establece las características de los
residuos peligrosos, el listado de los mismos y los límites que hacen a un
residuo peligroso por su toxicidad al ambiente, publicada en el Diario Oficial de la Federación
(D.O.F.) el 22 de octubre de 1993, la cual ha cambiado de nomenclatura en dos
ocasiones, la primera, por el Acuerdo Secretarial publicado en el D.O.F. el 29
de noviembre de 1994, siendo modificada a NOM-052-ECOL-1993 y, la segunda, por
el Acuerdo emitido en el mismo órgano de difusión el 23 de abril de 2003,
quedando con el nombre que aparece al inicio de esta cita.
3.5 Norma Oficial
Mexicana NOM-053-SEMARNAT-1993, Que establece el procedimiento para llevar a
cabo la prueba de extracción para determinar los constituyentes que hacen a un
residuo peligroso por su toxicidad al ambiente, publicada en el Diario Oficial de la Federación
(D.O.F.) el 22 de octubre de 1993, la cual ha cambiado de nomenclatura en dos
ocasiones, la primera, por el Acuerdo Secretarial publicado en el D.O.F. el 29
de noviembre de 1994, siendo modificada a NOM-053-ECOL-1993 y, la segunda, por
el Acuerdo emitido en el mismo órgano de difusión el 23 de abril de 2003,
quedando con el nombre que aparece al inicio de esta cita.
3.6 Norma Oficial
Mexicana NOM-054-SEMARNAT-1993, Que establece el procedimiento para determinar
la incompatibilidad entre dos o más residuos considerados como peligrosos por
la Norma Oficial Mexicana NOM-052-SEMARNAT-1993, publicada en el Diario Oficial de la Federación
(D.O.F.) el 22 de octubre de 1993, la cual ha cambiado de nomenclatura en dos
ocasiones, la primera, por el Acuerdo Secretarial publicado en el D.O.F. el 29
de noviembre de 1994, siendo modificada a NOM-054-ECOL-1993 y, la segunda, por
el Acuerdo emitido en el mismo órgano de difusión el 23 de abril de 2003,
quedando con el nombre que aparece al inicio de esta cita.
3.7 Norma Oficial
Mexicana NOM-085-SEMARNAT-1994, Contaminación atmosférica-Fuentes fijas-Para
fuentes fijas que utilizan combustibles fósiles sólidos, líquidos o gaseosos o
cualquiera de sus combinaciones, que establece los niveles máximos permisibles
de emisión a la atmósfera de humos, partículas suspendidas totales, bióxido de
azufre y óxidos de nitrógeno y los requisitos y condiciones para la operación
de los equipos de calentamiento indirecto por combustión, así como los niveles
máximos permisibles de emisión de bióxido de azufre en los equipos de
calentamiento directo por combustión, publicada en el
Diario Oficial de la Federación
(D.O.F.) el 2 de diciembre de 1994 como NOM-085-ECOL-1994, la cual cambió su
nomenclatura por el Acuerdo emitido en el D.O.F. el 23 de abril de 2003,
quedando como aparece al inicio de esta cita.
3.8 Norma Oficial
Mexicana NOM-087-SEMARNAT-SSA1-2002, Protección
ambiental-Salud ambiental- Residuos peligrosos biológico-Infecciosos-Clasificación
y especificaciones de manejo, publicada en el Diario Oficial de la Federación (D.O.F.) el 17 de febrero de 2003
como NOM-087-ECOL-SSA1-2002, la cual cambió su nomenclatura por el Acuerdo
emitido en el D.O.F. el 23 de abril de 2003, quedando como aparece al inicio de
esta cita.
3.9 Norma
Mexicana NMX-AA-009/1993-SCFI. Contaminación atmosférica-Fuentes
fijas-Determinación de flujo de gases en un conducto por medio del Tubo de
Pitot, publicada en el Diario Oficial de
la Federación del 27 de diciembre de 1993.
3.10 Norma
Mexicana NMX-AA-10-SCFI-2001, Contaminación Atmosférica.- Fuentes fijas.-
Determinación de la emisión de partículas contenidas en los gases que fluyen
por un conducto.- Método isocinético, publicada en el Diario Oficial de la Federación del 18 de abril de 2001.
3.11 Norma
Mexicana NMX-AA-23/1986, Protección al Ambiente. Contaminación Atmosférica.
Terminología, publicada en el Diario
Oficial de la Federación el 15 de julio de 1986.
3.12 Norma
Mexicana NMX-AA-035-1976.
Determinación de bióxido de carbono, monóxido de carbono y oxígeno en los gases
de combustión, publicada en el Diario
Oficial de la Federación el 10 de junio de 1976.
3.13 Norma
Mexicana NMX-AA-054-1978.
Contaminación atmosférica-Determinación del contenido de humedad en los gases
que fluyen por un conducto-Método gravimétrico, publicada en el Diario Oficial de la Federación el 2 de
agosto de 1978.
3.14 Norma
Mexicana NMX-AA-055-1979. Contaminación atmosférica-Fuentes fijas-Determinación
de bióxido de azufre en gases que fluyen por un conducto, publicada en el Diario Oficial de la Federación el 6 de
septiembre de 1979.
3.15 Norma
Mexicana NMX-AA-070-1980. Contaminación atmosférica-Fuentes fijas-Determinación
de cloro y/o cloruros en los gases que fluyen por un conducto, publicada en el Diario Oficial de la Federación el 8 de
septiembre de 1980.
3.16 Norma
Mexicana NMX-B-036-1981. Definiciones relativas al Carbón y Coque, publicada en
el Diario Oficial de la Federación
el 27 de enero de 1982.
3.17 Norma
Mexicana NMX-Z-13-02-1981. Guía para la redacción, estructuración y
presentación de las normas oficiales mexicanas, publicada en el Diario Oficial de la Federación el 14
de mayo de 1981.
3.18 Protocolo de
1996. Relativo al Convenio sobre la
Prevención de la Contaminación del Mar por vertimiento de desechos y otras
materias, 1972.
4.
Definiciones y terminología
Para efectos de esta Norma
Oficial Mexicana se consideran las definiciones contenidas tanto en la Ley
General del Equilibrio Ecológico y la Protección al Ambiente y en la Ley
General para la Prevención y Gestión Integral de los Residuos, así como en los
Reglamentos en materia de Residuos Peligrosos y Prevención y Control de la
Contaminación de la Atmósfera, y las siguientes:
4.1 Alimentación
de residuos
Suministro de residuos a la
cámara de combustión del incinerador.
4.2 Alimentación
automática
Carga de los residuos a la
cámara de combustión primaria del incinerador mediante mecanismos de clausura
hermética que operan a presión negativa.
4.3 Alimentación
manual
Carga de residuos realizada
por los operadores directamente a la cámara de combustión primaria del
incinerador.
4.4 Cámara de
combustión final
Compartimiento en donde se
lleva a cabo la combustión final de los gases producidos por la incineración de
los residuos.
4.5 Cámara de
combustión primaria
Compartimiento en donde se
realiza la ignición y se lleva a cabo la combustión de los residuos.
4.6 Capacidad
calorífica del equipo
Es la cantidad de calor de
diseño que requiere el equipo de incineración para mantener las condiciones de
operación durante una hora y sus unidades son Joules/h.
4.7 Combustión
Proceso controlado de
oxidación rápida que se sucede durante la combinación de oxígeno con aquellos
materiales o sustancias contenidas en los residuos capaces de oxidarse.
4.8 Congénere
Se refiere a un compuesto
particular que pertenece a la misma familia química.
4.9 Descarga de
aguas residuales
Acción de verter,
infiltrar, depositar o inyectar aguas residuales a un cuerpo receptor.
4.10 Emisión
La descarga a la atmósfera
de toda sustancia en cualquiera de sus estados físicos o de energía.
4.11 Equipo de
control de emisiones
Dispositivo de control
operado al final de los equipos de proceso y cuyo propósito es reducir al
mínimo la emisión de partículas y gases de combustión.
4.12 Equivalente
tóxico (EQT)
Forma de reporte de
resultados de los congéneres sustituidos en las posiciones 2,3,7,8 de las
Dioxinas y Furanos en el cual se estandarizan las concentraciones detectadas de
acuerdo a su toxicidad relativa a la de la 2,3,7,8 Tetraclorodibenzo-p-dioxina
(TCDD).
4.13 Incineración
Cualquier proceso para
reducir el volumen y descomponer o cambiar la composición física, química o
biológica de un residuo sólido, líquido o gaseoso, mediante oxidación térmica,
en la cual todos los factores de combustión como la temperatura, el tiempo de
retención y la turbulencia, pueden ser controlados, a fin de alcanzar la
eficiencia, eficacia y los parámetros ambientales previamente establecidos. En
esta definición se incluye la pirólisis, la gasificación y el plasma, cuando
los subproductos combustibles generados en estos procesos sean sometidos a
combustión en un ambiente rico en oxígeno.
4.14 Incinerador
Equipo empleado para la
oxidación térmica de residuos con o sin recuperación de calor producido por la
combustión, con sus respectivos dispositivos de control de temperatura y de
composición de gases, así como con tolvas para la recepción de cenizas.
4.15 Instalación
de incineración
Predio ocupado por las
unidades de incineración para la oxidación térmica de residuos, con o sin
recuperación del calor producido por la combustión, incluyendo las áreas de
recepción, almacenamiento y tratamiento previo de los residuos, el incinerador,
sus sistemas de alimentación de residuos, combustible y aire, los sistemas de
tratamiento de los gases de escape y de las aguas residuales, así como los
dispositivos y sistemas de control de las operaciones de incineración, registro
y supervisión de las condiciones de operación.
4.16 Instalación
de incineración existente
Cualquier instalación de
incineración autorizada por la Secretaría o no autorizada que se encuentre
operando con anterioridad a la publicación de esta Norma Oficial Mexicana.
4.17 Ley
Ley General del Equilibrio
Ecológico y la Protección al Ambiente.
4.18 Límite máximo
permisible
Valor asignado a un
parámetro, el cual no debe ser excedido en la emisión de contaminantes.
4.19 Monitoreo
continuo
El que se realiza con
equipo automático con un mínimo de 15 lecturas en un periodo no menor de 60
minutos y un periodo no mayor de 360 minutos. El resultado del monitoreo es el
promedio del periodo en el que se llevó a cabo el muestreo.
4.20 Operador
Calificado
Operador que demuestre
tener experiencia mínima de seis meses en el uso y operación de incineradores.
4.21 PROFEPA
La Procuraduría Federal de
Protección al Ambiente.
4.22 Protocolo de
Pruebas
Secuencia de actividades
para verificar la eficiencia del sistema, determinar el nivel de eficiencia de
destrucción alcanzado por los sistemas de combustión y de control de emisiones,
la confiabilidad de los sistemas de monitoreo continuo de emisiones y de los
procedimientos adecuados de manejo de los residuos y subproductos.
4.23 Residuos
industriales no peligrosos
Aquellos generados en
procesos industriales que no estén considerados en la Norma Oficial Mexicana
NOM-052-SEMARNAT-1993 y aquellos que la Secretaría certifique como tales.
4.24 Residuos
Sólidos Urbanos
Los generados en las casas
habitación, que resultan de la eliminación de los materiales que utilizan en
sus actividades domésticas, de los productos que consumen y de sus envases,
embalajes o empaques; los residuos que provienen de cualquier otra actividad
dentro de establecimientos o en la vía pública que genere residuos con
características domiciliarias, y los resultantes de la limpieza de las vías y
lugares públicos, siempre que no sean considerados como residuos de otra
índole.
4.25 Residuos
peligrosos
Aquellos residuos definidos
por la NOM-052-SEMARNAT-1993.
4.26 Residuos
peligrosos biológico-infecciosos
Aquellos
residuos así considerados en la NOM-052-SEMARNAT-1993 y la
NOM-087-SEMARNAT-SSA1-2002.
4.27 Responsable
de la Instalación de Incineración
Persona física o moral a
quien se extiende una autorización, en los términos de las disposiciones
legales aplicables, para llevar a cabo actividades de incineración.
4.28 Secretaría
Secretaría de Medio
Ambiente y Recursos Naturales (SEMARNAT).
4.29 Sistema de
monitoreo continuo de emisiones
Consiste en un dispositivo
de medición automático continuo para la determinación de la concentración de un
contaminante, reportado en horarios promedios móviles.
5.
Especificaciones
5.1 Los
responsables de la instalación de incineración de residuos peligrosos deben
presentar a la Secretaría un resultado del protocolo de pruebas dentro del
plazo señalado en su autorización.
En el caso de incineración
de residuos peligrosos, los resultados del protocolo de pruebas deben ser
presentados en los términos y formalidades que establece el Trámite
SEMARNAT-07-012 “Autorización para el manejo de residuos peligrosos que
pretendan su reuso, reciclaje, tratamiento o incineración”, del Acuerdo por el
que se dan a conocer los trámites inscritos en el Registro Federal de Trámites
y Servicios que aplica la Secretaría de Medio Ambiente y Recursos Naturales y
sus órganos administrativos desconcentrados y se establecen diversas medidas de
mejora regulatoria, publicado en el Diario
Oficial de la Federación el 29 de mayo de 2003.
5.2 Las instalaciones
de incineración deben operar en todo momento con un operador calificado en la
operación del equipo.
5.3 La
instalación de incineración debe contar con un sistema de registro de datos a
través de bitácoras o archivos electrónicos, aplicables a la recepción,
almacenamiento, proceso de incineración incluyendo los sistemas de control de
emisiones, monitoreo de contaminantes y disposición de residuos sólidos de
acuerdo a lo que establezca la Secretaría.
5.4 Las bitácoras
deben ser libretas foliadas, el registro también puede ser en archivos
electrónicos, en ambos casos deben guardarse por un tiempo mínimo de 5 años.
5.5 No debe
llevarse a cabo la incineración de residuos peligrosos que sean o contengan
compuestos orgánicos persistentes y bio-acumulables; plaguicidas
organoclorados; así como baterías y acumuladores usados que contengan metales
tóxicos; siempre y cuando exista en el país alguna otra tecnología disponible
que cause menor impacto y riesgo ambiental.
6.
Recepción de los residuos
6.1 Es requisito
indispensable para la instalación que presta servicios a terceros para la
aceptación de los residuos peligrosos, la presentación del Manifiesto de
Entrega-Transporte-Recepción de Residuos Peligrosos.
6.2 En el caso de
residuos peligrosos y de la instalación de incineración que presta servicios a
terceros, el responsable de la instalación de incineración, antes de aceptar el
ingreso de este tipo de residuos a su establecimiento, debe verificar:
a) Si la
composición física y química de los residuos peligrosos coincide con los
descritos por el generador en el Manifiesto y si éstos son compatibles con el
equipo de incineración;
b) La masa de
los residuos;
c) Las medidas
adecuadas para su almacenamiento y manejo conforme a las características de
incompatibilidad que, en su caso, puedan presentar respecto de otros residuos
peligrosos recibidos;
d) La empresa
habrá de efectuar una medición por radiación, utilizando un detector de
centelleo, en caso de que la lectura sea mayor a dos veces el fondo, se dará
aviso de inmediato a la Comisión Nacional de Seguridad Nuclear y Salvaguardias
y se procederá siguiendo las instrucciones que indique la misma.
e) En caso de no
satisfacer las condiciones mencionadas en el inciso a), los residuos peligrosos
no deben ser recibidos en la instalación del incinerador.
6.3 Para el caso
de los residuos considerados como no peligrosos, no es necesario cumplir con lo
indicado en los incisos anteriores.
7.
Operación de una instalación de incineración
7.1 La
instalación de incineración debe contar con un área de almacenamiento, de
conformidad con los ordenamientos jurídicos aplicables; para los materiales y
residuos, con una capacidad mínima de por lo menos dos veces la capacidad
diaria de operación autorizada.
7.2 La instalación
de incineración debe contar con los sistemas de control o con una planta
generadora de energía eléctrica para emergencias, que garanticen el paro seguro
y la combustión completa de los residuos en caso de falla del suministro
eléctrico.
7.3 La instalación
de incineración contará con un sistema para el pesaje de los residuos que se
reciban.
7.4 Las empresas
de servicios a terceros deben contar con un laboratorio dentro de sus
instalaciones, el cual deberá realizar una evaluación presuntiva del contenido de
cloro en cada lote de residuos admitidos, por cualquier método de análisis.
Esto no aplica para residuos biológico-infecciosos.
7.5 El diseño,
equipamiento y funcionamiento de las instalaciones de incineración deben
permitir que la temperatura de los gases derivados de la incineración de los
residuos se eleve, tras la última inyección de aire de combustión, de manera
controlada y homogénea, aun en las condiciones más desfavorables, hasta por lo
menos 850°C, alcanzados en o cerca de la pared interna, de la cámara de
combustión final, durante un tiempo mínimo de por lo menos dos segundos. En el
caso de que se incineren residuos peligrosos que contengan más del 1% de
sustancias organocloradas expresadas en cloro, la temperatura deberá elevarse
hasta 1,100°C, y durante 2 segundos como mínimo.
Cuando se compruebe que por
cuestiones tecnológicas, de eficiencia de los equipos y por la corriente de los
residuos a incinerar, la temperatura de operación pueda ser menor a 1100ºC, con
una eficienciade destrucción del 99.9999%
para el compuesto organoclorado de mayor estabilidad térmica que se encuentre
en dicha corriente de residuos, la Secretaría podrá autorizar la operación a
una temperatura inferior a la señalada en este inciso, misma que no podrá ser
menor a 850ºC y el tiempo de residencia será de dos segundos como mínimo.
Por el contrario, para
aquellos residuos cuya temperatura de destrucción sea mayor a 1100ºC, la
Secretaría podrá determinar la temperatura y tiempo de residencia a cumplir,
para garantizar su destrucción.
En el caso de los equipos
que incineren exclusivamente Residuos Peligrosos-Biológico Infecciosos (RPBI),
el tiempo de residencia puede ser menor a dos segundos, siempre y cuando se
cumpla con los límites de emisión que aparecen en la Tabla 1 de esta Norma
Oficial Mexicana.
7.6 Para evitar
las emisiones fugitivas, la presión de operación de las cámaras de combustión
del incinerador debe ser negativa.
7.7 La unidad de
incineración debe estar equipada con quemadores que se pongan en marcha de manera
automática cuando la temperatura descienda por debajo de la mínima establecida
para su operación.
7.8 La unidad de
incineración debe contar con un sistema de paro automático en la alimentación
de residuos peligrosos el cual se acciona cuando:
a) Durante la
puesta en marcha, no se alcance la temperatura mínima requerida;
b) No logre
mantenerse la temperatura mínima de incineración requerida;
c) Las emisiones
de monóxido de carbono (CO) sobrepasen los valores máximos permisibles;
7.9 El operador
debe mantener un registro diario en bitácora foliada o archivos electrónicos, a
disposición de la PROFEPA, en la cual registrará la siguiente información:
a) Tipo y
cantidad de residuos incinerados, en el caso de las empresas de servicios a
terceros, los resultados del análisis de cloro a que se refiere el párrafo 7.4
de la presente Norma;
b) Temperatura
del equipo en las diferentes cámaras y equipos de control;
c) Tipo y
cantidad de combustible consumido;
d) Arranques,
paros y horas de operación del equipo;
e) Fallas y
problemas presentados durante la operación del equipo, señalando las medidas
correctivas adoptadas para el restablecimiento de las condiciones normales de
operación;
f) Mediciones de
los contaminantes especificados en las tablas de esta Norma; en el caso de
mediciones continuas referenciar la localización de los registros;
g) Condiciones
de operación del equipo de control de emisiones (presión, temperatura y tasa de
alimentación);
h) Cantidad,
tipo y destino final de los residuos generados por el incinerador, y
i) Nombre y
firma del responsable de la instalación de incineración.
7.10 No se permite
la alimentación manual del incinerador; la alimentación con una carga de
residuos mayor o con residuos diferentes a los que han sido autorizados por la
Secretaría.
7.11 Las
instalaciones de incineración deben de contar con un Programa para Atención a
Contingencias y con los sistemas o procedimientos para prevenir y responder a
incendios o explosiones, así como a fugas o derrames de residuos.
7.12 Las cenizas y
otros residuos sólidos que se generen durante los procesos de incineración,
serán considerados como residuos peligrosos, por lo que su manejo deberá
cumplir con lo establecido en los ordenamientos legales aplicables.
7.13 Si el
contenido de materia volátil en la ceniza es mayor al 10%, éstas deben ser
realimentadas al incinerador.
7.14 Las descargas
de aguas residuales procedentes de las instalaciones de incineración, deben
cumplir con lo dispuesto en la normatividad aplicable o las condiciones
particulares de descarga que, en su caso, establezca la autoridad competente.
7.15 Las
instalaciones de incineración que cuenten con autorización para llevar a cabo
el tratamiento de bifenilos policlorados y compuestos organoclorados, según lo
establecido en el apartado 7.5,
deben demostrar anualmente una eficiencia de destrucción y remoción (EDR) de al
menos 99.9999 por ciento, respecto
al compuesto de mayor dificultad de destrucción presente en el residuo
alimentado.
Para determinar el
compuesto de mayor dificultad presente en el residuo alimentado, es necesario
tomar como base el compuesto organoclorado con mayor estabilidad térmica que se
encuentre en la corriente de residuos a incinerar.
El cálculo de la eficiencia
de destrucción y remoción está dado por la fórmula:
EDR
= Ai-Ei x 100% Ai
Ai = Flujo másico del
componente contenido en la alimentación al incinerador, calculado por el
producto de la concentración del componente de mayor dificultad de destrucción
en el residuo alimentado g/h.
Ei = Flujo másico del
componente de mayor dificultad de destrucción presente en el residuo alimentado
contenido en las emisiones a la atmósfera y las cenizas generadas.
Dicho flujo se calcula:
Ei = (Qi X G) + (mi X Mc)
Donde:
Qi = Concentración de la emisión del compuesto de mayor dificultad
de destrucción, g/m3.
G = Caudal del gas de emisión en la chimenea, m3/h.
mi = Concentración del componente de mayor dificultad de destrucción
en las cenizas, g/Kg.
Mc = Caudal de cenizas generadas, Kg/h.
En el caso de que por
cuestiones propias del proceso de incineración la cantidad de cenizas en los
fondos del incinerador sea despreciable, la variable Mc será cero y el segundo
término de la ecuación será despreciable.
7.16 En caso de
que por razones de fallas en los equipos de alimentación automática, medición
continua, control de emisiones, o alguna otra falla que impida el
funcionamiento de la operación autorizada del incinerador, se debe suspender la
alimentación de los residuos. La recepción de los mismos podrá continuar
siempre y cuando no se rebase la capacidad del área de almacenamiento de
acuerdo a lo establecido en el numeral 7.1, de la presente Norma Oficial
Mexicana.
8.
Medición en chimenea
8.1 La
instalación de incineración debe contar con sistemas para la medición continua
de indicadores de buenas prácticas de operación y control, contando por lo
menos con un equipo de monitoreo continuo para la temperatura de la cámara de
combustión final y para las emisiones de monóxido de carbono (CO) y oxígeno (O2),
a la salida de los gases de chimenea.
8.2 Para llevar a
cabo la medición de las emisiones a la atmósfera, los incineradores deben
contar con plataforma y puertos de muestreo en el ducto de salida de los gases
de acuerdo con lo establecido en el artículo 17 fracción III del Reglamento de
la Ley en materia de Prevención y Control de la Contaminación de la Atmósfera y
de acuerdo a lo especificado en la Norma NMX-AA-009/1993-SCFI, referida en el
punto 3 de esta Norma Oficial Mexicana.
9. Emisiones al
ambiente
9.1 Los límites
máximos permisibles de emisiones son los establecidos en la Tabla 1 de la
presente Norma Oficial Mexicana.
9.2 La
temperatura máxima de los gases antes del equipo de control de emisiones cuando
se utilicen lavadores secos debe ser menor a 250°C. En los demás casos, la
temperatura de los gases a la salida de la chimenea no debe rebasar dicho
valor.
9.3 Los límites
máximos permisibles de emisión, la frecuencia de medición y los métodos de
evaluación son los establecidos en la Tabla 1 de esta Norma Oficial Mexicana y
se aplicarán todo el tiempo para las instalaciones de incineración, excepto en
periodos de arranque o paro de los equipos.
9.4 En caso de
mal funcionamiento del equipo de medición continua, debe efectuarse al menos
una medición diaria puntual hasta que el desajuste sea corregido y dar aviso de
inmediato a la Secretaría, de la falla y el tiempo estimado para su ajuste,
para que ésta determine lo conducente. La utilización de métodos de evaluación,
distintos a los señalados en la Tabla 1 se sujetará a lo establecido en la Ley
Federal sobre Metrología y Normalización y su Reglamento.
En caso de que no sea
posible llevar a cabo la medición diaria puntual se debe suspender la
alimentación de los residuos al incinerador.
9.5 El responsable
de la instalación de incineración podrá quedar exento de realizar el análisis
de alguno o varios de los parámetros establecidos en la Tabla 1 de esta Norma
Oficial Mexicana, cuando demuestre a la Secretaría que por las características
de los residuos a tratar no genera o concentra los contaminantes a exentar,
manifestándolo a ella por escrito y bajo protesta de decir la verdad. En caso
de falsedad, el responsable queda sujeto a los ordenamientos legales
aplicables.
La
disposición anterior no aplica para los parámetros relacionados con la calidad
de la combustión (CO, NOx).
Estas exenciones sólo
podrán ser autorizadas por un plazo no mayor a 2 años, siempre y cuando los
resultados de 3 años consecutivos de mediciones de los parámetros a exentar estén
25% por debajo de los límites máximos permisibles indicados en la Tabla 1.
TABLA 1
LIMITES
MÁXIMOS PERMISIBLES DE EMISIONES PARA INSTALACIONES
DE
INCINERACIÓN DE RESIDUOS
|
CONTAMINANTE |
LIMITE DE EMISIÓN |
FRECUENCIA DE MEDICIÓN |
NORMA QUE APLICA O MÉTODO |
|
CO (mg/m3) |
63 |
CONTINUO |
Infrarrojo No Dispersivo
y Celda Electroquímica Anexo 1 |
|
HCl (mg/m3) |
15 |
TRIMESTRAL |
NMX-AA-070-1980 |
|
NOx (mg/m3) |
300 |
SEMESTRAL |
Quimiluminiscencia Anexo 2 |
|
SO2 (mg/m3) |
80 |
SEMESTRAL |
NMX-AA-55-1979 |
|
PARTÍCULAS (mg/m3) |
50 |
SEMESTRAL |
NMX-AA-10-SCFI-2001 |
|
ARSÉNICO SELENIO COBALTO NÍQUEL MANGANESO ESTAÑO (mg/m3) |
0.7* |
SEMESTRAL |
Espectrometría de
absorción atómica. Anexos 3 y 4 |
|
CADMIO (mg/m3) |
0.07 |
SEMESTRAL |
Espectrometría de
absorción atómica. Anexos 3 y 4 |
|
PLOMO CROMO total COBRE ZINC (mg/m3) |
0.7* |
SEMESTRAL |
Espectrometría de
absorción atómica. Anexos 3 y 4 |
|
MERCURIO (mg/m3) |
0.07 |
SEMESTRAL |
Espectrometría de
absorción atómica con vapor frío Anexos 3 y 4 |
|
DIOXINAS Y FURANOS EQT
(ng/m3) Instalaciones de
incineración nuevas |
0.2 |
ANUAL |
Cromatografía de gases
acoplado a espectrometría de masas de alta resolución Anexo 5A |
|
DIOXINAS Y FURANOS EQT
(ng/m3) Instalaciones de
incineración existentes antes de la publicación de esta NOM. |
0.5 |
ANUAL |
Cromatografía de gases
acoplado a espectrometría de masas de baja resolución Anexo 5B |
Todos los valores están
referidos a condiciones estándar: 1 atmósfera, base seca, 25ºC y 7% de Oxígeno
O2, de acuerdo a la NOM-085-SEMARNAT-1994.
* Suma total metales
pesados.
**Todas las mediciones
deben estar registradas en bitácora.
10. Evaluación de
la conformidad
La Secretaría reconocerá
las determinaciones analíticas que hayan sido muestreadas y analizadas por un
laboratorio acreditado y aprobado conforme a las disposiciones legales
aplicables.
11. Grado de
concordancia con normas y lineamientos internacionales y con las normas
mexicanas tomadas como base para su elaboración
Esta Norma Oficial Mexicana
no concuerda con ninguna norma o lineamiento internacional, tampoco existen
normas mexicanas que hayan servido de base para su elaboración.
12. Bibliografía
12.1 Aseguramiento
de la Calidad/Control de la Calidad Procedimientos para la Incineración de
Residuos Peligrosos EPA/625/6-89/0.
12.2 Exposure of man to dioxins: a perspective on industrial waste
incineration. ISBN-8072-49.
Reporte
Técnico No. 49 (Exposición humana a dioxinas: una perspectiva sobre la
incineración de residuos industriales).
12.3 LaGrega
Michael, Buckingham Phillip L., y Evans Jeffrey C. "Gestión de residuos
tóxicos", Tratamiento, eliminación y recuperación de suelos. Vols. I y II, McGraw-Hill, México, 1996.
12.4 Martínez
Mondragón Jaime y Ortiz Monasterio Fernando (ERM-México, S.A. de C.V.)
"Tecnología de control de Dioxinas y Furanos y un caso de estudio de
muestreo", presentado en el Seminario Internacional sobre Incineración,
México, agosto, 1998.
12.5 Método 23 de
la EPA.- "Determinación de policlorodibenzodioxinas y
policlorodibenzofuranos provenientes de fuentes estacionarias".
12.6 Standards of Performance for New Stationary Sources: Medical Waste
Incinerators, 27 de febrero de 1995 [Estándares de Desempeño para Fuentes Fijas
Nuevas: Incineradores de Residuos Médicos].
12.7 Strong Brian and Copland Richard. "Summary of the Final New Source
Performance Standards and Emission Guidelines for New and Existing
Hospital/Medical/Infectious Waste Incinerators". Presentado en
la 91a. Reunión Anual de la Air & Waste Management Association del 14 al 16
de junio de 1998 en San Diego, California, EUA pp. 9.
12.8 Tchobanoglous
George, Theisen Hilary y Vigil Samuel A., "Gestión integral de residuos
sólidos", Vol. I y II, McGraw-Hill, México, 1996.
12.9 Van Ruymbeke
Claire. “Propuesta de Norma de Emisiones a la atmósfera por la incineración de
Residuos Hospitalarios”. Informe interno de distribución restringida para la
Dirección General de Materiales, Residuos y Actividades Riesgosas del Instituto
Nacional de Ecología. SEMARNAP, México, 1997.
12.10 Directiva de
la Comunidad Europea 2000/76/EC, relativa a la incineración de residuos. Parlamento Europeo. 28 de diciembre de 2000.
12.11 Taylor, Phillip H., Barry Dellinger, and C. C.
Lee (University of Dayton and USEPA), "Development of a thermal
stability-based ranking of hazardous organic compound incinerability",
Environmental Science and Technology. Vol. 24; Pág. 316-328. Marzo, 1990.
13. Observancia de la norma
La Secretaría a través de
la Procuraduría Federal de Protección al Ambiente, los gobiernos de los
estados, del Distrito Federal y de los municipios, en el ámbito de sus
respectivas competencias y atribuciones, vigilarán el cumplimiento de la
presente Norma Oficial Mexicana. El incumplimiento de la presente Norma Oficial
Mexicana será sancionado conforme a lo dispuesto en la Ley General del
Equilibrio Ecológico y la Protección al Ambiente, la Ley General para la
Prevención y Gestión Integral de los Residuos, y demás ordenamientos jurídicos
aplicables.
TRANSITORIOS
PRIMERO.- La presente
Norma Oficial Mexicana entrará en vigor 60 días después de su publicación en el
Diario Oficial de la Federación.
SEGUNDO.- Los límites
máximos permisibles de emisión de dioxinas y furanos en incineradores
existentes serán revisados quinquenalmente por la Secretaría y el Grupo de
Trabajo de esta Norma Oficial Mexicana, tomando en consideración aspectos
ambientales y de salud pública, de desarrollo tecnológico y la conveniencia de
aprovechar la vida útil de las instalaciones existentes.
TERCERO.- A la
entrada en vigor del Plan Nacional derivado de la obligatoriedad del Convenio
de Estocolmo se revisarán las especificaciones aplicables a los equipos de
monitoreo, particularmente de aquellos que determinen las emisiones de dioxinas
y furanos, en los incineradores nuevos.
México, Distrito Federal, a
los cinco días del mes de agosto de dos mil cuatro.- El Subsecretario de Fomento
y Normatividad Ambiental de la Secretaría de Medio Ambiente y Recursos
Naturales y Presidente del Comité Consultivo Nacional de Normalización de Medio
Ambiente y Recursos Naturales, Juan Rafael Elvira Quesada.- Rúbrica.
ANEXO 1
ESPECIFICACIONES Y
PROCEDIMIENTOS DE PRUEBA PARA SISTEMAS
DE MONITOREO CONTINUO EN EMISIONES (SMCE) DE MONOXIDO DE CARBONO (CO)
INDICE
1. Objetivo y campo de aplicación
2. Alcance y principio
3. Definiciones
4. Interferencias
5. Seguridad
6. Equipo y accesorios
7. Reactivos y materiales de referencia
8. Procedimiento de evaluación de
desempeño
9. Control de calidad
10. Calibración y trazabilidad
11. Procedimiento de análisis
12. Cálculos
13. Especificaciones para pruebas de
desempeño
14. Procedimiento alterno para la prueba de
exactitud relativa
15. Ejemplos de pruebas de desempeño
16. Bibliografía
1. Objetivo y
campo de aplicación
1.1 Objetivo.
Establecer las
Especificaciones y Procedimientos de Prueba requeridos para un Sistema de
Monitoreo Continuo de Emisión (SMCE) utilizado para la determinación continua
de la concentración (relación analito/matriz) de Monóxido de Carbono (CO) en
los gases que fluyen en un ducto.
1.2
Aplicabilidad.
Estas Especificaciones
aplican de manera obligatoria para Sistemas de Monitoreo Continuo de Emisiones
(SMCE) de Monóxido de Carbono (CO).
Estas Especificaciones
aplican para SMCE del tipo Extractivo o Estacionario.
Estas Especificaciones no
están diseñadas para evaluar el desempeño de SMCE por periodos prolongados de
tiempo, ni identifica técnicas específicas de calibración requeridas para
evaluar su desempeño. El dueño de la fuente emisora y su operador, son los
responsables de calibrar, mantener y operar de manera apropiada el SMCE.
Se podrá solicitar al dueño
de la fuente emisora y operador, la evaluación del desempeño del SMCE en
cualquier momento distinto a la prueba inicial.
2. Alcance y
principio
2.1 Alcance.
Se establecen las
Especificaciones y Procedimientos de Prueba para SMCE de:
Analito: Monóxido de
Carbono (CO).
Número CAS: 630-80-0.
2.2 Principio.
Las presentes
Especificaciones incluyen:
- Especificaciones de
Instalación y Medición
- Especificaciones de
Desempeño
- Procedimientos de Prueba
- Manejo de Datos
Se utilizan métodos de
referencia, pruebas de error de calibración, pruebas de desplazamiento de
calibración y pruebas de interferencia para evaluar el grado de conformidad con
las presentes Especificaciones.
3. Definiciones
3.1 Sistema de Monitoreo
Continuo de Emisiones (SMCE).
Es el
total del equipo requerido para la determinación de la concentración de un
analito o conjunto de analitos en una matriz gaseosa que pasa de manera
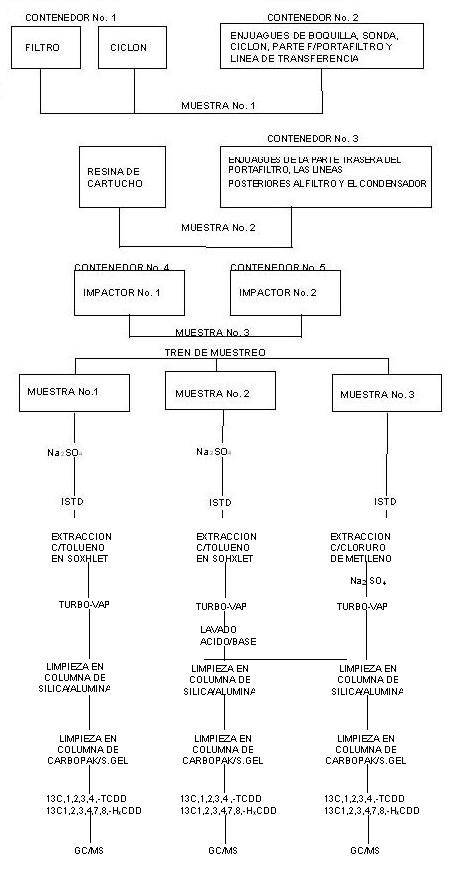
continua por el elemento sensor. El Sistema
de Monitoreo se conforma de 4 principales subsistemas: (1) Interfase de
Muestra; (2) Analizador; (3) Dilutor, y(4) Registrador.
Los SMCE son divididos en
dos categorías generales: (1) Extractivos, y (2) Estacionarios.
3.1.1 SMCE Extractivo.
Aquellos
que extraen muestra del ducto, la transportan, la acondicionan y la analizan.
En ocasiones el acondicionamiento consiste en enfriamiento, eliminación de
humedad por distintos mecanismos y eliminación de sólidos. Aquellos componentes
de un Sistema de Monitoreo que realizan estas funciones forman parte de la
Interfase de Muestra. En ocasiones se utilizan mecanismos de dilución
controlada de muestra para ajustar la concentración del analito a magnitudes
propias de detección. Los SMCE extractivos que no utilizan sistema de dilución
se denominan del tipo Extracción Completa.
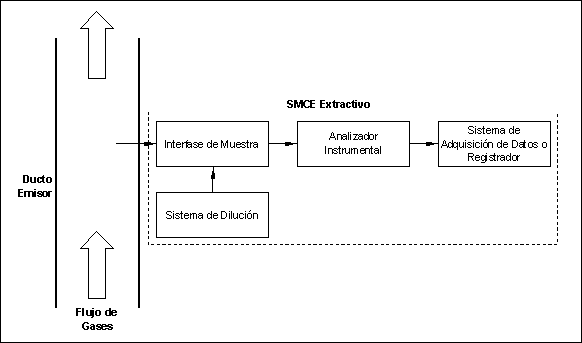
Figura 1.- Subsistemas de un SMCE Extractivo.
3.1.2 SMCE Estacionarios
(De Detección en Sitio).
Aquellos
que realizan la detección y cuantificación del analito dentro del ducto emisor
y sin necesidad de extraer o tomar parte de la muestra. Existen dos tipos
generales de SMCE Estacionarios: (1) Puntuales, y (2) de Paso.
3.1.2.1 SMCE Estacionario
Puntual.
Aquel
sistema que consiste de un sensor electro-químico o electro-óptico montado en
la punta de una sonda que se introduce en el ducto. Cuando la detección se
realiza con un sensor electro-óptico, la trayectoria de paso deberá ser menor o
igual a un 10% del diámetro equivalente del ducto o chimenea para que sea
considerado un SMCE Estacionario Puntual (de lo contrario ver SMCE Estacionario
de Paso).

Figura
2.- Diagrama de SMCE Estacionario Puntual.
3.1.2.2 SMCE Estacionario
de Paso.
Los SMCE Estacionarios de Paso evalúan el analito a lo largo de un trayecto dentro del ducto (y no en un solo punto). Utilizan sensores electro-ópticos. Son sistemas que detectan y cuantifican el analito mediante la emisión de un haz de luz a lo largo de un trayecto dentro del ducto, y la interacción entre el analito y este haz de luz es utilizada para cuantificarlo. Los SMCE Estacionarios de Paso varían en función de la cantidad de veces en que un mismo haz de luz se hace pasar a través del trayecto del ducto seleccionado. Para que un SMCE sea considerado Estacionario de Paso, la trayectoria de paso deberá ser mayor a un 10% del diámetro equivalente del ducto o chimenea, sin embargo, en la presente Especificación se requiere que el 70% trayectoria de paso esté dentro del 50% del área transversal del ducto.
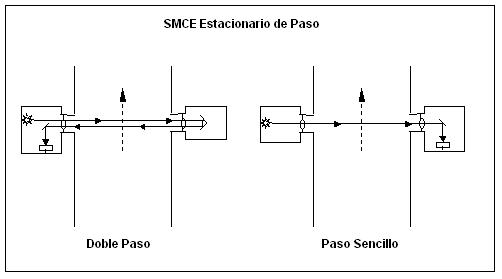
Figura 3.-
Diagrama de SMCE Estacionario de Paso.
3.2 Desplazamiento de
Calibración.
La
diferencia entre la respuesta inicial del SMCE al calibrarlo inicialmente con
un valor de referencia, y su respuesta al alimentar el mismo valor de
referencia después de un determinado periodo de muestreo u operación sin llevar
a cabo calibraciones intermedias, mantenimiento, reparación y/o apagado del
sistema de muestreo.
3.3 Área Centroidal.
Es el área concéntrica que
es geométricamente similar a la sección transversal del ducto o chimenea.
3.4 Sistema de Adquisición de Datos o Registrador.
Parte o subsistema del SMCE
que registra permanentemente los datos del analizador instrumental. Este
sistema de adquisición de datos o registrador puede poseer capacidad de
reducción de datos automática.
3.5 Sistema de Dilución.
Parte o subsistema del SMCE
que utiliza diluye de manera controlada y cuantificada la muestra que es
alimentada al analizador con el fin de reducir la concentración del analito a
magnitudes propias de detección del analizador instrumental. El gas utilizado
para diluir no interacciona con el gas muestreado y el elemento sensor.
3.6 Analizador
Instrumental.
Parte o subsistema del SMCE
que detecta y cuantifica el analito o grupo de analitos.
3.7 Exactitud
Relativa (ER).
Es
la diferencia absoluta promedio entre la concentración del analito determinada
por el SMCE y aquella determinada por el Método de Referencia, más un
coeficiente obtenido con un 97.5% de
confianza de una serie de pruebas y dividido por el promedio de concentración
obtenido en las distintas pruebas usando el Método de Referencia, o por el
límite de emisión aplicable a la fuente.
3.8 Interfase de Muestra.
Parte
o subsistema del SMCE utilizada para cualquiera de las siguientes funciones:
(1) adquisición de muestra; (2) transporte de muestra; (3) acondicionamiento de
muestra, y (4) protección del SMCE del efecto del efluente en el ducto.
3.9 Límite Superior de
Medición o Registro (LSMR).
Límite superior de
concentración de cada rango o intervalo de medición del sistema de adquisición
de datos, registrador y/o analizador de gases.
Un SMCE de rango sencillo
con un analizador instrumental capaz de detectar y cuantificar hasta 1000 ppmv
de un analito, pero con un sistema de adquisición de datos o registrador, capaz
de registrar información hasta 500 ppmv, posee un LSMR de 500 ppmv.
Un SMCE de rango sencillo
con un analizador instrumental capaz de detectar y cuantificar hasta 1000 ppmv
de un analito, pero con un sistema de adquisición de datos o registrador, capaz
de registrar información hasta 5000 ppmv, posee un LSMR de 1000 ppmv.
Un SMCE de multirrango
capaz de detectar, cuantificar y registrar en rangos de 0 a 100 con precisión
de 0.1, de 0 a 1 000 con precisión
de 1 y de 0 a 10 000 con precisión de 5, su LSMR varía en función al rango
seleccionado durante el muestreo, siendo el LSMR de 100, 1 000 y 10 000 para
cada rango, respectivamente.
3.10 Tiempo de Respuesta (TR).
Tiempo requerido para que
el SMCE registre un 95% de un cambio escalonado de concentración de gas.
Figura
4.- Diagrama de Tiempo de Respuesta.
3.11 Error de
Calibración (EC).
Es la diferencia entre la
concentración del SMCE y la concentración generada por la fuente calibración,
cuando el gas de calibración es alimentado al SMCE. La evaluación del EC es
utilizada para evaluar la exactitud y linealidad del SMCE en todo su rango de
medición.
3.12 SMCE Rango
Sencillo.
Aquel SMCE en el que el
Analizador Instrumental y el Sistema de Adquisición de Datos o Registrador,
poseen sólo un rango de medición de concentración. Este rango va generalmente
de cero al LSMR del rango sencillo.
3.13 SMCE
Multirrango.
Aquel SMCE en el que el
Analizador Instrumental y el Sistema de Adquisición de Datos o Registrador,
poseen dos o más rangos de medición de concentración. Estos rangos van
generalmente de cero o del LSMR del rango anterior, al LSMR del rango
siguiente, y por lo general la sensibilidad del instrumento disminuye a medida
que el rango aumenta.
3.14 Muestreo
Multipuntual Integral.
Se refiere a la toma de
muestra en un intervalo de tiempo considerable a flujo constante, suficiente
para obtener una muestra integral de un volumen mayor o igual a 30 litros. La
toma de la muestra se realiza en varias ubicaciones definidas dentro de un
corte transversal del ducto (varios puntos transversales de muestreo).
3.15
Estratificación
Cuando la concentración de
un analito en un corte transversal del ducto varía significativamente en
distintos puntos dentro del ducto. La estratificación se considera
significativa cuando la concentración promedio dentro del ducto y la
concentración en cualquier punto transversal ubicado a una distancia mayor a 1
metro de la pared del ducto, es mayor a un 10%.
4.
Interferencias
No establecidas
5.
Seguridad
Los procedimientos
solicitados en estas Especificaciones pueden involucrar el uso de materiales,
operaciones y equipos peligrosos. Estas Especificaciones pueden omitir la
indicación de riesgos asociados a los procedimientos indicados, por lo que será
responsabilidad del usuario el establecer prácticas de operación seguras antes
de realizar estos procedimientos. Se recomienda consultar el manual de usuario
del SMCE para tomar las debidas precauciones.
6.
Equipo y accesorios
6.1
Especificaciones para el SMCE.
6.1.1 Escala del
Sistema de Adquisición de Datos o Registrador.
El rango de salida del
Sistema de Adquisición de Datos o Registrador deberá incluir los valores de
cero y alto nivel. El SMCE deberá ser capaz de medir los niveles de
concentración bajo condiciones normales de operación del proceso, así como los
picos de alta concentración de corta duración. Este doble rango de medición
podrá ser logrado utilizando dos analizadores independientes de rango sencillo
(uno de bajo y otro de alto rango de medición), o utilizando analizadores de
multirrango de medición (un solo analizador con la capacidad de medir en dos o
más rangos distintos). En el caso de analizadores multirrango, cuando la
concentración detectada llegue y rebase el LSMR del rango inferior o uno
intermedio, la operación del siguiente rango de medición deberá entrar de
manera automática. Bajo aplicaciones en las que la concentración del analito
sea consistentemente baja, se puede utilizar un SMCE de rango sencillo siempre
y cuando se logren detectar y cuantificar los picos de alta concentración. En
este caso se deberá seleccionar un valor apropiado de alto nivel que incluya
estos picos.
Para SMCE de multirrango,
los rangos inferior y superior deberán poseer las siguientes características:
|
Rango |
LSMR |
|
Inferior |
LSMR = 200 ppmv |
|
Superior |
LSMR = 3000 ppmv |
Tabla
1
Especificaciones para el
LSMR en SMCE Multirrango.
SMR (=) Límite Superior de
Medición o Registro
Deberá preverse que no
existan intervalos de concentración entre dos rangos inmediatos que no sean
evaluados. Por ejemplo, no es admisible que un SMCE de dos rangos posea un
rango de mediciónde 0 a 200 ppmv y el siguiente de 1 000 a 3 000 ppmv.
Si se utiliza un
Registrador análogo, el registro de datos se deberá establecer de manera que el
valor de alto nivel quede entre un 90 y un 100% de la escala máxima del
Registrador. Este requerimiento puede ser inaplicable a Sistemas de Adquisición
de Datos o Registradores digitales.
La división real de escala
del Sistema de Adquisición de Datos o Registrador deberá ser equivalente a 0.5% del LSMR.
6.1.2 El diseño del
SMCE deberá permitir la determinación del desplazamiento de calibración en los
valores de cero y alto nivel. Si lo anterior no es posible o fuese impráctico,
el diseño deberá permitir la determinación del desplazamiento de calibración en
un valor entre el cero y el equivalente al 20% del valor de alto nivel, y a un
valor entre el 50 y el 100% del valor de alto nivel. En casos especiales, se
podrá autorizar la evaluación del desplazamiento de calibración en un solo
valor de concentración.
6.1.3 Frecuencia de
Medición y Almacenamiento o Registro de Datos.
El SMCE deberá de ser capaz
de medir la concentración del analito al menos una vez cada 15 segundos. Una
concentración promedio de estas lecturas debe calcularse y registrarse en al
menos cada 15 minutos.
6.1.4 Cálculo del
Promedio Móvil Horario.
El SMCE debe calcular un
Promedio Móvil Horario al menos cada 15 minutos, el cual es el promedio
aritmético de los 4 valores más recientes de los registros de cada 15 minutos.
6.2 Otros Equipos
y Accesorios.
Aquellos requeridos por el
Método de Referencia.
7.
Reactivos y materiales de referencia
7.1 Gases Patrón,
Celdas de Gas Patrón y/o Filtros Opticos.
Utilizar los Gases Patrón,
Celdas de Gas Patrón y/o Filtros Opticos especificados por el fabricante del
SMCE.
7.2 Reactivos y
otros Materiales de Referencia.
Aquellos requeridos por el
Método de Referencia.
8.
Procedimiento de evaluación de desempeño
8.1
Especificaciones de Instalación y Sitio de Muestreo.
8.1.1 Instalación
del SMCE.
Instale el SMCE en un lugar
accesible en el que la concentración del analito sea directamente
representativa o pueda ser corregida de manera que represente el total de la
emisión del proceso evaluado.
La ubicación óptima de la
Interfase de Muestra de un SMCE Extractivo es determinada por varios factores,
incluyendo la facilidad de acceso para efectuar las operaciones de calibración
y mantenimiento, el grado de acondicionamiento requerido para la muestra, el
nivel de representatividad respecto a la emisión total, y el grado con que se
representan las situaciones reales de emisión.
Seleccione puntos de
medición o trayectorias de paso representativas en sitios en que el SMCE pase
la prueba de Exactitud Relativa (ver más adelante). Si la causa de que el SMCE
no apruebe la prueba de Exactitud Relativa es el sitio de muestreo
seleccionado, y resulta inaplicable una técnica de corrección de resultados, se
podrá solicitar la reubicación del SCME en otro sitio de muestreo. En el
siguiente inciso se indican distintos sitios de muestreo que propician la
instalación apropiada.
8.1.2 Sitio de
Muestreo para el SMCE en un Corte Longitudinal del Ducto.
Se sugiere que el sitio de
muestreo en un corte longitudinal del ducto sea:
(1) al menos a una distancia equivalente a
dos diámetros equivalentes de ducto recto sin perturbaciones después de una
perturbación, algún dispositivo de control, el punto donde se genera el analito
o cualquier otro punto en el que la concentración del analito cambie, y
(2) al menos a una distancia equivalente a
medio diámetro equivalente de ducto recto sin perturbaciones de la salida del
efluente gaseoso o de la entrada al dispositivo de control.
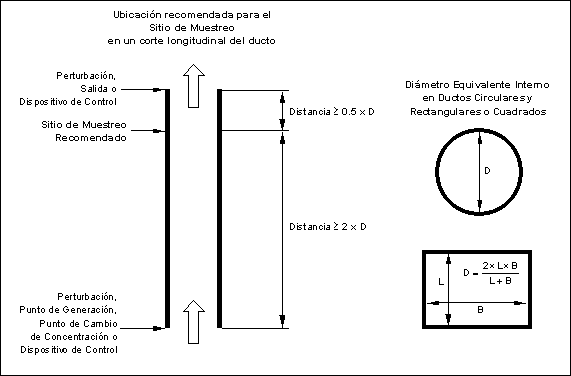
Figura
5.- Ubicación recomendada para el Sitio de Muestreo en un corte
longitudinal del ducto
El Sitio de Muestreo
corresponde a la ubicación física del SMCE
8.1.2 Sitio de
Muestreo para el SMCE en un Corte Transversal del Ducto.
8.1.2.1 Punto de
Muestreo para SMCE Extractivo o Estacionario Puntual.
Se sugiere que el punto de
muestreo en un corte transversal del ducto sea:
(1) a una distancia mayor o igual a 1 metro
de la pared del ducto, o
(2) centrado dentro del ducto.
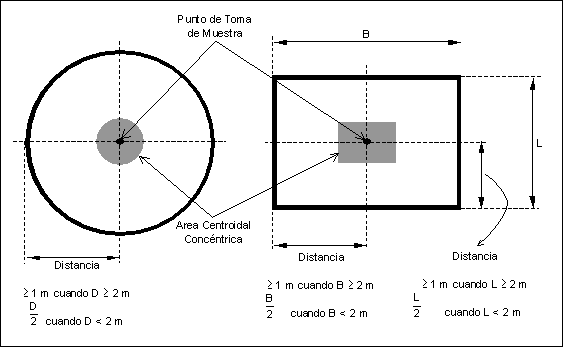
Figura
6.- Ubicación recomendada para el punto de muestreo en un corte transversal
del ducto
8.1.2.2 Trayectoria
de Medición para SMCE Estacionario de Paso.
Se sugiere que la
trayectoria de medición efectiva en un corte transversal del ducto sea:
(1) totalmente dentro de un
área concéntrica delimitada por una línea a 1 metro de las paredes del ducto;
(2) que el 70% de la trayectoria de paso
esté dentro del 50% del área transversal del ducto, o
(3) esté localizada dentro de cualquier
parte del área concéntrica del ducto.
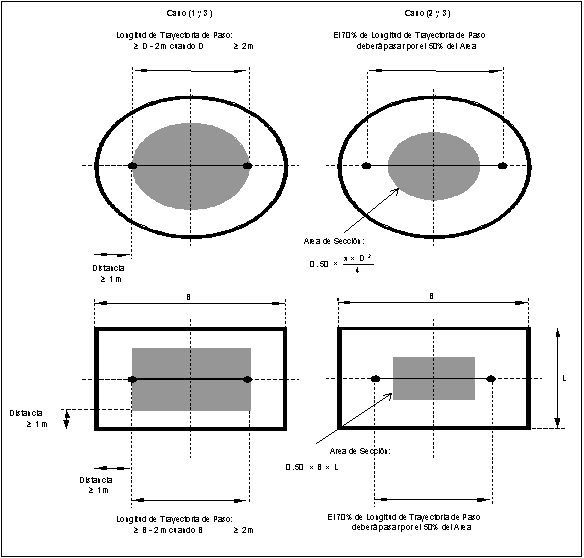
Figura
7.- Trayectoria de Medición efectiva recomendada en corte transversal del
ducto
8.1.3 Criterios
para Ubicación de SMCE en Función a otros Sistemas de Monitoreo Continuo o
Intermitentes.
Cuando se instalen otros
Sistemas de Monitoreo Continuos o Intermitentes para la medición de parámetros
o compuestos necesarios para la corrección de los resultados generados por el
SMCE, como son el O2 o CO2 para corrección por dilución,
o el contenido de humedad para la corrección de resultados de concentración
medidos en base húmeda[1] a
base seca, ambos sistemas deberán ser instalados en el mismo sitio de muestreo.
En caso de no ser posible, estos sistemas podrán ser instalados en sitios de
muestreo distintos siempre y cuando estos sitios no estén estratificados y que
no existan fugas que permitan la dilución de los gases entre ambos sitios.
8.1.4 Sitio de
Muestreo y Puntos Transversales para la Prueba por Método de Referencia.
8.1.4.1 Seleccione un
punto de muestreo que esté ubicado en:
(1) al menos a una distancia equivalente a
dos diámetros equivalentes de ducto recto sin perturbaciones después de una
perturbación, algún dispositivo de control, el punto donde se genera el analito
o cualquier otro punto en el que la concentración del analito cambie, y
(2) al menos una a una
distancia equivalente a medio diámetro equivalente de ducto recto sin
perturbaciones de la salida del efluente gaseoso o de la entrada al dispositivo
de control.
Cuando
los cambios de concentración del contaminante se atribuyen únicamente a
dilución por fugas (por ejemplo, fugas en un recuperador de calor); se puede
usar como criterio de ubicación medio diámetro equivalente en vez de los dos
diámetros equivalentes requeridos.
La
ubicación del sitio de muestreo del SMCE y del Método de Referencia no tiene
que ser la misma.
8.1.4.2 Seleccione puntos
transversales que aseguren la adquisición de muestras representativas en el
área transversal del ducto. Los requerimientos mínimos son los siguientes:
(a) establezca una “línea de medición” que pase por el centro y en dirección
a cualquier posible estratificación;
(b) si esta línea interfiere con la medición del SMCE, desplace
la línea 30 centímetros o una distancia equivalente al 5% del diámetro
equivalente del ducto, la que resulte menor;
(c) ubique tres puntos transversales a 16.7,
50.0 y 83.3% de la línea de medición;
(d) si la línea de medición es mayor a 2.4 metros y no se espera estratificación, los puntos podrán ser
fijados a 0.4, 1.2 y 2.0 metros de la pared interna del ducto (esta opción no
podrá ser utilizada después de lavadores húmedos o en puntos en donde se unen
dos corrientes con distintas concentraciones del analito), y
(e) aquellos puntos transversales que resulten a una distancia
menor a 3 centímetros de la pared, deberán ser ajustados a 3 centímetros de la
pared.
8.1.4.3 Procedimiento
de Prueba de Estratificación.
Para determinar si existe
estratificación en el efluente, se utiliza un sistema de dos sondas. Una sonda
es ubicada en el centro del ducto y es utilizada únicamente para determinar si
existen cambios significativos de concentración en el proceso (sonda de
“referencia”), mientras que la otra es utilizada para determinar la concentración
del gas en 12 puntos transversales seleccionados de acuerdo a la NMX-AA-009
(sonda de “muestreo”), respetando la distancia de 3 centímetros desde la pared
(criterio indicado en el inciso anterior). Determine la concentración en cada
punto transversal (Ci) registrando adicionalmente la concentración
registrada simultáneamente en la sonda de referencia (CREF,i).
Calcule la diferencia
absoluta entre ambos valores para cada punto transversal con la siguiente
ecuación:
![]()
Ecuación 1
Donde:
C*i = Diferencia
de concentraciones corregidas por la referencia para cada punto transversal
número “i”.
Ci = Concentración registrada en punto
transversal número “i”.
CREF,i = Concentración en sonda de referencia durante
la medición en el punto transversal número “i”
Calcule la concentración
promedio en el ducto:
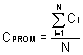
Ecuación 2
Donde:
CPROM = Concentración promedio en el
ducto.
Ci = Concentración
registrada en punto transversal número “i”.
N = Número total de puntos transversales.
Determine si existe
estratificación calculando la diferencia absoluta en forma porcentual y
comparándola contra el criterio de rechazo de 10%:
Si ![]() > 10 entonces existe estratificación.
> 10 entonces existe estratificación.
La siguiente tabla presenta
un ejemplo del cálculo para la prueba de estratificación.
|
Punto
Transversal No.
“i” |
Ci (ppmv) |
CREF,I (ppmv) |
C*I (ppmv) |
(%) |
Se
Detecta Estratificación |
|
1 |
102 |
100 |
2 |
1.38 |
NO |
|
2 |
98 |
92 |
6 |
4.13 |
NO |
|
3 |
93 |
91 |
2 |
1.38 |
NO |
|
4 |
142 |
158 |
16 |
11.01 |
SI |
|
5 |
165 |
165 |
0 |
0.00 |
NO |
|
6 |
142 |
141 |
1 |
0.69 |
NO |
|
7 |
142 |
143 |
1 |
0.69 |
NO |
|
8 |
143 |
143 |
0 |
0.00 |
NO |
|
9 |
125 |
122 |
3 |
2.06 |
NO |
|
10 |
178 |
183 |
5 |
3.44 |
NO |
|
11 |
201 |
200 |
1 |
0.69 |
NO |
|
12 |
213 |
203 |
10 |
6.88 |
NO |
|
Promedio |
CPROM = 145.33 |
|
|
|
|
Tabla
2
Ejemplo numérico de la
Prueba de Estratificación
Cuando exista
estratificación, esto es cuando uno o más de los puntos transversales así lo
indiquen, se deberá omitir utilizar el criterio 0.4, 1.2 y 2.0 metros en ductos
con diámetro equivalente mayor a 2.4 metros, y se deberá utilizar en estos
casos el criterio de 16.7, 50.0 y 83.3% sobre la línea de medición.
Se pueden utilizar otros
puntos transversales de muestreo bajo previa autorización.
8.2 Preparativos
para Prueba.
Instale el SMCE y prepare
el sistema de medición para el Método de Referencia conforme a las indicaciones
anteriores.
8.3 Procedimiento
de Prueba para la Evaluación del Desplazamiento de Calibración (DC).
8.3.1 Periodo de
Prueba para DC.
Mientras el proceso esté
operando bajo condiciones normales de operación o con cargas mayores o iguales
a un 50% de su carga normal de operación, determine el desplazamiento en la
calibración en intervalos de 24 horas durante 7 días consecutivos, haciendo uso
de los procedimientos indicados en los dos siguientes incisos.
8.3.2 El propósito
de la prueba de Desplazamiento de Calibración es demostrar la habilidad del
SMCE para mantener su calibración durante un periodo determinado de tiempo. Por
esto, es necesario que cuando el SMCE posea calibración periódica de manera
automática o manual, las pruebas de Desplazamiento de Calibración se deberán
efectuar antes de estos ajustes, o llevadas a cabo de manera que se evalúe el
Desplazamiento de manera apropiada.
8.3.3 Realice la
prueba de Desplazamiento de Calibración en los dos niveles de concentración
especificados en el inciso 6.1.2.
Introduzca al SMCE los Gases Patrón, Celdas de Gas Patrón o Filtros Ópticos
certificados. Registre el valor de respuesta del SMCE y calcule el Coeficiente
de Desplazamiento para cada nivel en el que se haya realizado la prueba (cero y
alto nivel).
![]() Ecuación 3
Ecuación 3
Donde:
DCi = Coeficiente de Desplazamiento de Calibración
obtenido para el Gas Patrón “i” o Celda Patrón “i” o Filtro Óptico “i”.
Ci = Respuesta del SMCE ante la alimentación
del Gas Patrón “i” o Celda Patrón “i” o Filtro Óptico “i”, después de 24 horas.
CPATRON,i = Concentración del analito en el Gas
Patrón “i” o Celda Patrón “i” o Filtro Óptico “i”.
LSMR = Límite Superior de Medición o
Registro.
Nota: se deberá ser
congruente con las unidades de concentración usadas para Ci, CPATRON,i
y LSMR.
8.4 Procedimiento
de Prueba para la Evaluación de la Exactitud Relativa (ER).
8.4.1 Periodo de
Prueba para ER.
Realice la prueba ER
mientras el proceso esté operando bajo condiciones normales de operación o con
cargas mayores o iguales a un 50% de su carga normal de operación. La prueba ER
puede llevarse a cabo de manera simultánea a la prueba DC.
8.4.2 Método de
Referencia para la Determinación de Monóxido de Carbono en los Gases que Fluyen
en un Ducto.
8.4.2.1 Objetivo,
Principio y Aplicabilidad
8.4.2.1.1 Objetivo.
Determinar la concentración
de monóxido de carbono (CO) en los gases que fluyen en un ducto.
8.4.2.1.2 Principio.
Una muestra gaseosa es
extraída del ducto de manera integral o continua de una corriente gaseosa.
Parte de la muestra es transportada a un analizador instrumental (o varios)
para el análisis del contenido de monóxido de carbono (CO). Los principios
instrumentales comúnmente utilizados para la detección del CO son la absorción
de radiación en la banda infrarroja (rayo infrarrojo no dispersivo), y
potenciometría (la diferencia de potencial generado en una celda
electroquímica).
8.4.2.1.3
Aplicabilidad.
Este método es aplicable
para la determinación de la concentración de monóxido de carbono en condiciones
diluidas (generalmente determinado en ppmv).
8.4.2.2 Rango y
Sensibilidad.
8.4.2.2.1 Rango.
El rango es determinado por
el diseño del Analizador Instrumental así como su Sistema de Adquisición de
Datos, Indicador o Registrador. Para este método, la parte del rango es
asignada por la selección del LSMR del sistema de medición. Se recomienda que
el LSMR del sistema sea seleccionado de manera que la concentración promedio de
CO no sea menor a un 20% del valor del LSMR. Si en algún caso el LSMR es
rebasado por la concentración de gas muestra, la medición será inválida.
El rango mínimo requerido
es de 0 a 1 000 ppmv.
8.4.2.2.2 Sensibilidad.
La sensibilidad máxima
aceptable para un analizador de 0 a 1 000 ppmv (LSMR = 1 000 ppmv) es de 20
ppmv (equivalente a 2% del LSMR).
8.4.2.3 Normas de
Referencia.
No aplica.
8.4.2.4 Definiciones
y Nomenclatura.
Igual a inciso 3.
8.4.2.5
Interferencias.
Para el caso de un
analizador por Rayo Infrarrojo No Dispersivo, cualquier sustancia que absorba
energía en la banda infrarroja puede llegar a interferir hasta cierto alcance.
Por ejemplo, para equipos que midan en un rango de 1500 y 3000 ppmv de CO, las
relaciones de discriminación para el agua (H2O) y bióxido de carbono
(CO2) son 3.5% H2O
por 7 ppmv CO y 10% CO2 por 10 ppmv CO, respectivamente. Para
equipos que midan en un rango de 0 a 100 ppmv, las relaciones de interferencias
pueden ser tan altas como 3.5% H2O
por 25 ppmv CO y 10% CO2 por 50 ppmv CO. El uso de acondicionadores
de muestra y filtro de correlación de gases, son comúnmente utilizados con este
tipo de analizadores.
Para analizadores por celda
electroquímica, el gas que comúnmente genera interferencia con el CO es el
hidrógeno molecular (H2), por lo que se debe de procurar no utilizar
este principio de medición en sistemas con alto contenido de este gas (por
ejemplo sistemas con gases de reducción). Existen celdas electroquímicas en el
mercado que poseen una relativa compensación por la presencia del hidrógeno.
8.4.2.6
Especificaciones de Desempeño del Sistema de Medición.
8.4.2.6.1 Error de
Calibración (EC).
Deberá ser menor a ± 2% del
LSMR para calibraciones de cero, niveles medio y alto.
8.4.2.6.2 Desviación
por Interfase de Muestra.
Deberá ser menor a ± 5% del
LSMR para calibraciones de cero, niveles medio y alto.
8.4.2.6.3
Desplazamiento de Cero.
Deberá ser menor a ± 3% del
LSMR para el periodo de tiempo de cada corrida (para Infrarrojo No Dispersivo
se permite un 10% en 8 h).
8.4.2.6.4
Desplazamiento de Calibración (Valor de Alto Nivel).
Deberá ser menor a ± 3% del
LSMR para el periodo de tiempo de cada corrida (para Infrarrojo No Dispersivo
se permite un 10% en 8 h).
8.4.2.7 Equipo.
8.4.2.7.1 Sistema de
Muestreo Integral.
8.4.2.7.1.1 Sonda.
Fabricada de acero
inoxidable o vidrio borosilicato. Para eliminar la carga de partículas del gas,
la sonda podrá poseer un filtro dentro o fuera del ducto (por ejemplo, un tapón
de fibra de vidrio, acero inoxidable poroso, etc.). Se podrá utilizar algún otro
material para la fabricación de la sonda, siempre y cuando éste sea inerte a
los gases presentes en el ducto, y que sea resistente a altas temperaturas.
8.4.2.7.1.2 Condensador o
Acondicionador de Gases.
Un condensador enfriado por
aire, agua o cualquier otro mecanismo (por ejemplo, placa de resistencia
eléctrica). Capaz de eliminar la humedad del gas a un nivel en que ya no
interfiera en la operación de la bomba, rotámetro e instrumento de medición.
8.4.2.7.1.3 Válvula.
Para regular el flujo de
gas se requiere de una válvula de aguja o equivalente.
8.4.2.7.1.4 Bomba.
Para transportar el gas a
la bolsa flexible se requiere de una bomba de diafragma o equivalente, la cual
deberá ser hermética. Se podrá colocar un tanque de supresión para evitar el
efecto pulsante de la bomba sobre el rotámetro. Cuando se utilice un tren de
muestreo en que el gas pase por la bomba, ésta no podrá utilizar agua, aceite o
algún otro fluido que entre en contacto con el gas muestreado.
8.4.2.7.1.5 Rotámetro.
Un rotámetro o equivalente,
capaz de medir flujo dentro de un 2%. Se recomiendan flujos entre 500 y 1000
cm³/min.
8.4.2.7.1.6 Bolsa
Flexible.
Una bolsa hermética hecha de plástico (i.e., Tedlar, Mylar, PTFE) o
plástico con cubierta de aluminio (i.e., Mylar Aluminizado), o equivalente.
Deberá tener una capacidad de almacenamiento acorde al flujo de gas y tiempo
total de muestreo seleccionados. Se recomiendan capacidades entre 55 y 90
litros. Para realizar una prueba de hermeticidad en la bolsa, conecte un
manómetro de columna de agua y presurice la bolsa a una presión de 5 a 10 cm de
H2O (2 a 4" H2O). Mantenga la bolsa así durante 10
minutos. Cualquier desplazamiento en la columna indicará fuga(s) en la bolsa.
Como procedimiento alterno se puede realizar un presurizando de igual manera
(sin incluir el manómetro), y esperar por una noche; si la bolsa se desinfla,
entonces existe fuga.
8.4.2.7.1.7 Manómetro.
Para pruebas de
hermeticidad en bolsas flexibles se utilizará un manómetro tipo "U"
con columna de 30 cm. H2O. Para pruebas de hermeticidad en el tren
de muestreo se utilizará un manómetro tipo "U" con columna de 76 cm
de Hg.

Figura
8.- Sistema de Muestreo Integral apropiado para Método de Referencia para CO
Algunos de los elementos
del diagrama no son estrictamente requeridos o pueden ser reemplazados por
elementos equivalentes.
8.4.2.7.2 Sistema de
Medición Continua.
Cualquier sistema de
medición para CO que cumpla con las especificaciones dictadas en este método
(ver siguiente Figura).
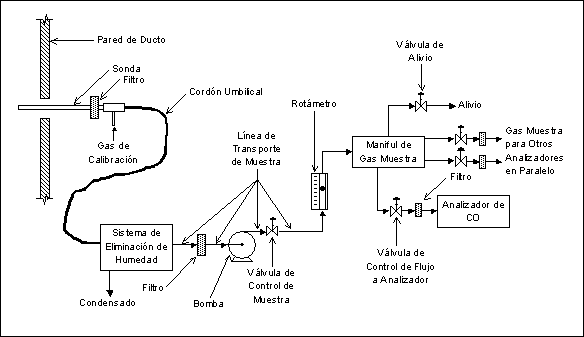
Figura
9.- Sistema de Muestreo Continuo apropiado para Método de Referencia para
CO
Algunos de los elementos
del diagrama no son estrictamente requeridos o pueden ser reemplazados por
elementos equivalentes.
8.4.2.7.2.1 Sonda.
Igual a Sonda de Sistema de
Muestreo Integral.
8.4.2.7.2.2 Analizador
Instrumental.
Un analizador capaz de
determinar la concentración de CO en forma continua. El analizador deberá
cumplir con las especificaciones de desempeño dictadas en el inciso 8.4.2.6 de este Método de Referencia.
Se deberá asignar un dispositivo de medición de flujo apropiado para el control
del flujo alimentado al analizador (por ejemplo, rotámetro). En caso de que el
analizador sea insensible al flujo volumétrico, el dispositivo de flujo puede
ser eliminado del sistema.
8.4.2.7.2.3 Trampa de
Bióxido de Carbono.
Como aditamento adicional
cuando se utilice el principio de medición por Infrarrojo No Dispersivo sin
filtro de correlación de gases, se podrá utilizar una trampa o tubo que
contenga ascarita (adsorbedor de CO2) ubicado después del
acondicionador de muestra (sistema de eliminación de humedad) con el fin de
reducir los posibles efectos de interferencia. Cuando se utilice esta trampa,
los resultados de CO deberán corregirse en función al contenido de CO2
en los gases.
8.4.2.7.2.4 Gases Patrón
para Calibración.
Los gases de calibración
para analizadores de CO, deberán ser CO balance N2. Para el gas de calibración cero deberá utilizar
nitrógeno de alta pureza. Se recomienda que la concentración del gas utilizado
para la calibración de alto nivel no exceda por 1.5 veces el límite máximo permisible de emisión del ducto
evaluado. Adicionalmente se recomienda que el gas de calibración de alto nivel
se encuentre en un valor aproximado (pero siempre por debajo) del LSMR del equipo.
También, se recomienda poseer mezclas con concentraciones de CO equivalentes a
30 y 60% el LSMR cuando el principio instrumental utilizado sea conocido por
tener desviaciones de linealidad (por ejemplo, Rayo Infrarrojo No Dispersivo).
De los criterios anteriores, se da mayor prioridad al criterio de 1.5 veces el Límite de Emisión, sin
embargo, este valor no podrá ser mayor al LSMR del sistema de medición.
8.4.2.7.2.5 Condensador o
Acondicionador de Gases.
Igual a Sonda de Sistema de
Muestreo Integral.
8.4.2.8 Procedimiento
para la Evaluación del Desempeño del Sistema de Medición.
8.4.2.8.1 Error de
Calibración (EC).
Instale el equipo en un
lugar seguro y de acuerdo al sistema de medición mostrado en la Figura 8 o 9 o
algún sistema análogo propio para el muestreo. Asegúrese de que el sitio de
instalación no posea en exceso humedad, vibraciones, ruido y fenómenos que
puedan dañar tanto al sistema de medición como al personal de muestreo.
Encienda los equipos, ajústelos y espere al menos el tiempo indicado en el
manual del equipo analizador para que éste se encuentre acondicionado y listo
para operar. Realice la siguiente prueba de hermeticidad: tapando la entrada al
sistema de medición, se deberá verificar que el flujo volumétrico disminuya a
cero en la entrada al analizador, o bien, aprobando la prueba de desempeño para
Desviación por Interfase de Muestra. Calibrar el analizador en la secuencia que
el responsable técnico considere apropiada, y con la alimentación de los gases
directamente al analizador instrumental (sin hacerlos pasar por la Interfase de
Muestra).
Calcule el Error de
Calibración para cada Gas de calibración utilizado, con la siguiente ecuación:
![]()
Ecuación 4
Donde:
EC = Error de
Calibración, (%)
CA = Concentración de respuesta del analizador al introducir el
Gas Patrón directo al analizador instrumental (una vez efectuados todos los
ajustes en el instrumento).
CPATRON = Concentración
Real del Gas Patrón.
LSMR = Límite Superior de
Medición o Registro.
Nota: se deberá ser
congruente con las unidades de concentración usadas para CA, CPATRON,
y LSMR.
Nota: el Error de Calibración (EC)
se evalúa para cada Gas de Calibración utilizado (cero, nivel medio y alto).
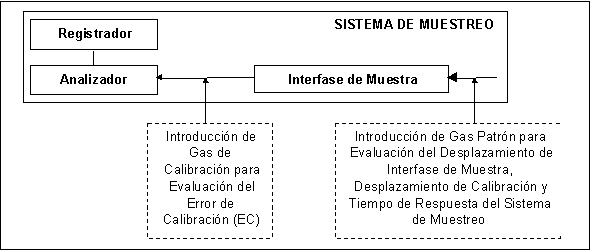
Figura
10.- Ubicación de Sitios para introducción del Gas Patrón para la evaluación
del Desempeño del Sistema Instrumental.
Criterio de Aceptación: EC
para cada Gas Patrón deberá ser menor a ± 2% para aceptar los resultados.
8.4.2.8.2 Desviación
por Interfase de Muestra (DIM).
Se utilizan únicamente dos
gases de calibración para esta prueba: (1) el Cero, y (2) el nivel Medio o
Alto, dependiendo de cuál posea la concentración más cercana a la concentración
del gas muestra. Introduzca el gas de calibración de Alto (o Medio) Nivel en la
punta de la sonda de muestreo o en la punta del umbilical (la sonda puede ser
excluida del sistema para esta prueba), y registre la concentración mostrada
por el sistema. Introduzca el gas de calibración Cero y registre la
concentración mostrada por el analizador. Durante la ejecución de esta prueba,
el sistema de muestreo deberá operar en la forma normal de muestreo, no realice
ningún cambio en el sistema de muestreo a menos que sean aquéllos necesarios
para obtener un apropiado flujo volumétrico de gas de calibración a la entrada
del analizador. En caso de existir algún tipo de fuga significativa (mala
instalación, cordón umbilical roto, etc.), ésta generará un rechazo por
desviación significativa. La fuga deberá corregirse y volverse a efectuar la
prueba.
Efectúe esta prueba antes
de comenzar el muestreo y al terminarlo.
Calcule la Desviación por
Interfase de Muestra con la siguiente ecuación:
![]() Ecuación 5
Ecuación 5
![]() Ecuación
5
Ecuación
5
Donde:
DIMINICIAL = Desviación por Interfase de Muestra Inicial,
(%).
DIMFINAL = Desviación
por Interfase de Muestra Final, (%).
CA = Concentración de respuesta del analizador al introducir el
Gas Patrón directo al analizador instrumental (una vez efectuados todos los
ajustes en el instrumento).
CSI = Concentración de respuesta del analizador al introducir el
Gas Patrón por la Interfase de Muestra al inicio del Muestreo.
CSF = Concentración de respuesta del analizador al introducir el
Gas Patrón por la Interfase de Muestra al término del Muestreo.
LSMR = Límite Superior de
Medición o Registro.
Nota: ser
congruente con las unidades de concentración usadas para CA, CSI,
CSF, y LSMR.
Nota: la Desviación
por Interfase de Muestra (DIM) Inicial y Final se puede evaluar para cada Gas
de Calibración utilizado (cero, nivel alto o medio).
Durante la ejecución de la
prueba de DIM, el responsable técnico podrá en forma opcional, determinar el
tiempo de respuesta del sistema de la siguiente manera: cuando se efectúe la
prueba del gas de calibración de Cero y Nivel Alto (o Medio), se mide el tiempo
que toma el analizador en alcanzar el 95% de la respuesta estable desde el
momento en que se introdujo el gas al sistema, y el tiempo mayor encontrado
entre ambos gases (Cero y Nivel Alto o Medio), será considerado como el tiempo de
respuesta del sistema.
Criterio de Aceptación: las
DIM Inicial y Final deberán ser menor a ± 5% para aceptar los resultados.
8.4.2.8.3
Desplazamiento de Calibración (DC).
Con los resultados de la
prueba de DIM, calcule el Desplazamiento de Calibración con la siguiente
ecuación:
![]()
Ecuación 5
Donde:
DC = Desplazamiento
de Calibración, (%).
CSI = Concentración de respuesta del analizador al introducir el
Gas Patrón por la Interfase de Muestra al inicio del Muestreo.
CSF = Concentración de respuesta del analizador al introducir el
Gas Patrón por la Interfase de Muestra al término del Muestreo.
LSMR = Límite Superior de
Medición o Registro.
Nota: ser
congruente con las unidades de concentración usadas para CSI, CSF,
y LSMR.
Nota: el Desplazamiento
se evalúa para cada Gas de Calibración utilizado (cero y nivel alto o medio).
Criterio de Aceptación: el
DC para cada Gas Patrón deberá ser menor a ± 3% para aceptar los resultados.
SEGUNDA
SECCIÓN
SECRETARIA DE
MEDIO AMBIENTE Y RECURSOS NATURALES
8.4.2.9 Procedimiento
de Muestreo.
8.4.2.9.1 Muestreo
Integral Puntual.
El punto de muestreo deberá
ser ubicado en el centro de un corte transversal del ducto, o bien, cuando el
ducto posea un diámetro equivalente mayor a 2 metros, en un punto de muestreo
puede ser ubicado a una distancia mayor o igual a 1 metro de distancia de la
pared del ducto.
Realice la prueba de
hermeticidad en la bolsa flexible. Antes de comenzar, realice una prueba de
hermeticidad del tren de muestreo: (1) coloque un manómetro de vacío en la
entrada al condensador, entre la sonda y el condensador; (2) conecte la bolsa
flexible y todas las partes del tren de muestreo; (3) tapela entrada a la sonda
y encienda la bomba hasta que se alcance un vacío mínimo de 250 mm Hg; (4)
apague la bomba y (5) el vacío deberá permanecer estable por un mínimo de 30
segundos. Una vez realizada y aprobada la prueba de hermeticidad, vaciar la
bolsa flexible por completo, conectarla de nuevo e introducir la sonda al punto
de muestreo. Como prueba alterna, se puede tapar la entrada de la sonda y
esperar a que el rotámetro baje a cero mientras la bomba de succión se
encuentre operando, si el rotámetro se mantiene en cero totalmente inmóvil, el
tren de muestreo se considera hermético desde la sonda al rotámetro. Después,
tapar la alimentación a la bolsa y si el rotámetro se mantiene totalmente
inmóvil, el tren de muestreo se encuentra hermético hasta la entrada a la
bolsa.
Extraer la muestra a flujo
constante. El flujo volumétrico indicado por el rotámetro deberá ser registrado
en intervalos de 1 a 5 minutos. Como prueba de calidad se deberán someter los
datos de flujo a:
Ecuación 6
Donde:
%Desviación =
Desviación de Flujo Promedio para la lectura de Flujo en el intervalo de toma
de lectura número. “i” (%).
Qi = Flujo
volumétrico de muestreo en el intervalo de toma de lectura número “i”.
QPROM = Flujo
volumétrico promedio (aritmético) en todo el muestreo.
Nota: ser
congruente con las unidades de flujo usadas para Qi y QPROM.
Criterio de Aceptación: %
DESVIACIONi deberá ser menor o igual a 10% en cada intervalo “i”,
para que el muestreo integral sea válido.
Se recomienda que el
volumen recolectado de muestra exceda de 30 litros, sin embargo, muestras
menores son aún aceptables. Una vez colectada la muestra, su análisis deberá
ser efectuado dentro de las primeras 8 horas.
Antes de hacer el análisis,
realice la prueba de hermeticidad para el analizador instrumental, calíbrelo y
evalúe su desempeño conforme a las indicaciones de este Método.
Introduzca la muestra
colectada al analizador y registre la Concentración de CO promedio.
8.4.2.9.2 Muestreo
Integral Multipuntual.
Seleccione el mínimo número
de puntos de muestreo, su arreglo y su ubicación en el corte transversal del
ducto, de acuerdo a la NMX-AA-009.
Realice el procedimiento de
muestreo descrito en la sección 8.4.2.9.1
de este Método de Referencia, con la excepción de que la muestra deberá ser
tomada en todos los puntos transversales de muestreo mediante un ajuste
periódico de la sonda a cada punto transversal durante el muestreo. Se deberá
muestrear por un intervalo de tiempo igual en cada punto. Y finalmente realice
el análisis de CO con el procedimiento en la sección 8.4.2.9.1 de este
método.
8.4.2.9.3 Muestreo
Continuo.
Seleccione el tiempo total de muestreo así como el intervalo de tiempo a utilizar para toma de resultados. El muestreo se puede realizar en un solo punto como se indica en la sección 8.4.2.9.1, o de manera multipuntual (sección 8.4.2.9.2). Ajuste los flujos volumétricos del sistema de manera que queden en forma igual a los utilizados en la prueba de DIM, o en caso de no haberse realizado esta prueba, de manera que el flujo de alimentación de muestra al analizador, quede en forma igual al flujo de gas de calibración utilizado durante la calibración del analizador. Una vez instalado y funcionando, permita que pase un tiempo equivalente a 5 veces el tiempo de respuesta del sistema de medición (en caso de haberse evaluado), o bien, permita que el sistema llegue a una respuesta lógica y de relativa estabilidad. Arranque el cronómetro y registre la lectura inicial del analizador (tiempo cero). Posteriormente, registre en cada intervalo de tiempo la lectura del analizador, hasta cumplir el tiempo total de muestreo. Durante el mismo, verificar que los flujos volumétricos del sistema de medición no varíen conforme a los establecidos inicialmente. En caso de haberse realizado un muestreo multipuntual, indicar en cada intervalo de registro, el punto de muestreo.
En cuanto a los tiempos de
muestreo, utilizar las siguientes relaciones:
|
Tiempo Total de Muestreo |
|
Intervalo Máximo de Toma
de Lectura |
|
Menor a 1 hr |
|
1 min. o un mínimo de 30
lecturas (el que resulte en un
menor número de lecturas) |
|
Mayor o Igual a 1 hr |
|
2 min. o un mínimo de 96
lecturas (el que resulte en un
menor número de lecturas) |
El intervalo de tiempo
utilizado para la toma de lectura deberá ser constante a lo largo de todo el
muestreo.
En caso de haberse
utilizado el tubo de ascarita para la eliminación de CO2 (solicitado
para sistemas de Infrarrojo No Dispersivo sin filtro de correlación de gases),
evaluar el contenido de este compuesto en forma simultánea, con el fin de poder
corregir la concentración de CO por el volumen eliminado. Otra forma de evaluar
el contenido de CO2 cuando se utiliza el tubo de ascarita es: (1)
pesar el tubo de ascarita antes del muestreo; (2) registrar el volumen de
muestra extraído durante el muestreo ya sea integrando el flujo volumétrico del
rotámetro y/o por algún otro dispositivo de evaluación de volumen de gases, y
(3) evaluar el peso final del tubo de ascarita. Mediante el peso ganado por la
ascarita (CO2) y el volumen de muestreado, determinar el contenido
de CO2 aproximado en la muestra.
8.4.2.10 Cálculos.
8.4.2.10.1 Concentración
Promedio.
Para muestreos integrales,
la concentración promedio corresponde al resultado indicado por el analizador
al alimentar la muestra capturada.
Para muestreos continuos la
concentración promedio de CO en la muestra corresponde al promedio aritmético
de las lecturas obtenidas.
Para fines de conversión
entre unidades típicas de concentración de CO, utilice la siguiente conversión:
1 ppmv de CO = 1.144287 mg/m³ de CO, a Condiciones
Normales (298ºK y 101325 Pa).
8.4.2.10.2 Correcciones.
Para todos los analizadores
de CO, la concentración promedio mostrada por el analizador es corregida con el
DIM promedio obtenido para el Sistema de Medición con la siguiente ecuación:
![]()
Ecuación 7
Donde:
CGAS = Concentración de CO en el efluente gaseoso.
CPROM = Concentración
promedio de CO indicada por el analizador (ver inciso
8.4.2.10.1 de este método).
C0 = Concentración promedio de CO indicada por el analizador
durante el Chequeo Inicial y Final del
DIM de la Interfase de Muestra con el Gas de Calibración Cero.
CM = Concentración promedio de CO indicada por el analizador
durante el Chequeo Inicial y Final del
DIM de la Interfase de Muestra con el Gas de Calibración de Nivel Alto (o
Medio).
CPATRON = Concentración
real del Gas de Calibración de Nivel Alto (o Medio), ppmv, base seca.
Nota: ser
congruente con las unidades de concentración usadas.
En caso de haberse
utilizado la Trampa de Ascarita para Adsorber el CO2.
![]()
Ecuación 7
Donde:
CGAS = Concentración de CO en el efluente gaseoso.
CPROM = Concentración
promedio de CO indicada por el analizador (ver inciso
8.4.2.10.1 de este
método).
%VCO2 = Concentración
promedio de CO2 en la muestra gaseosa durante el muestreo (%
volumen).
Nota: se deberá ser
congruente con la referencia de humedad entre CPROM y %VCO2.
8.4.2.11 Registro.
Diseñe una hoja de campo
que solicite como mínimo la siguiente información:
- Empresa Propietaria de la Fuente Evaluada
- Fuente Evaluada
- Características de la Fuente Evaluada
- Identificación de los Elementos del Sistema de
Medición Utilizado
- Fecha y Hora de Inicio y Terminación
- Pruebas de Hermeticidad
- Gases Patrón Utilizados
- Calibración
- Error de Calibración
- Desviación de la Interfase de Muestra
- Desplazamiento de Calibración
- Tiempo Total de Muestreo
- Intervalo de Toma de Lectura
- Concentración de CO en cada Intervalo de
Lectura
- Identificación del Responsable del Muestreo
- Observaciones del Responsable del Muestreo
8.4.3 Estrategia de
Muestreo para Pruebas por Método de Referencia (MR).
Realice las pruebas con MR
de manera que se obtengan resultados representativos de la emisión del proceso
y que se puedan correlacionar con los resultados del SMCE de manera directa o
mediante correcciones apropiadas. Preferiblemente realice mediciones
simultáneas al MR de aquellos parámetros o compuestos necesarios para la
corrección de los resultados generados por el MR y SMCE, como son el O2 o
CO2 para corrección por dilución (si es aplicable), o el contenido
de humedad para la corrección de resultados de concentración medidos en base
húmeda a base seca (en caso de ser necesaria). Sin embargo, cuando se midan
parámetros o compuestos para corrección dentro de un periodo de 1 hora en que
fue determinada la concentración del analito, la corrección con estos
resultados es permitida. Para poder correlacionar los resultados del SMCE y MR
de manera apropiada, se deberá registrar la cronología de cada prueba efectuada
por MR y SMCE (inicio y terminación), incluyendo el horario exacto del día.
8.4.3.1 Muestras
Integrales por MR.
Si se utiliza el procedimiento de Muestreo Integral, el tiempo total de muestreo deberá ser de al menos 21 minutos por muestra, y cambie los puntos de muestreo del MR por los puntos indicados en el inciso 8.1.4.
8.4.3.2 Muestreo Continuo
por MR.
Si se utiliza el
procedimiento de Muestreo Continuo, el tiempo total de muestreo deberá ser de
al menos 60 minutos por corrida.
8.4.4 Número de
Muestras o Corridas de Prueba por MR.
Se requiere un mínimo de 9
muestras integrales o 9 corridas por muestreo continuo. En el caso en que
alguna de las corridas o muestras sea rechazada por control de calidad, se
deberá de repetir la corrida o muestra. Finalmente se requiere un mínimo de 9
pares de datos generados por MR y SMCE para realizar la prueba de ER.
8.4.5 Correlación
de Resultados MR y SMCE.
Correlacionar los
resultados del MR con los obtenidos por el SMCE correspondientes al mismo
periodo de muestreo. Los resultados corresponden al promedio de concentración
obtenido en cada corrida o muestra por MR y el promedio integrado de
concentración reportado por SMCE durante la toma de cada muestra o corrida del
MR. Cuando sea significativo, considere los tiempos de respuesta de cada
sistema para correlacionar los resultados que ambos sistemas obtuvieron
simultáneamente. Para cada par de resultados, confirme que sean consistentes
las correcciones de humedad y dilutor (O2 o CO2). Compare
cada par de resultados usando las siguientes indicaciones.
8.4.5.1 Si el MR fue
aplicado por muestreo integral, haga una comparación directa entre el promedio
integrado del SMCE.
8.4.5.2 Si el MR fue
aplicado por muestreo continuo, calcule el resultado de concentración promedio
mediante el promedio aritmético de todas las lecturas registradas y haga una
comparación directa con el promedio integrado del SMCE. Como procedimiento
alterno, registre la respuesta del SMCE durante cada intervalo de lectura del
MR, y compare ambos promedios aritméticos.
8.4.6 Calcule la
diferencia promedio para cada par de resultados MR-SMCE, exprésela en las
unidades de concentración del límite máximo de emisión del ducto evaluado.
Calcule la Desviación Estándar (Sd), el Coeficiente de Confianza
(CC) y la Exactitud Relativa (ER), haciendo uso de las indicaciones de la
sección de cálculos.
8.5 Procedimiento
de Prueba para Error de Calibración (EC).
Una vez calibrado el SMCE,
sométalo a tres distintos gases patrón (cero, nivel medio y alto), con
concentraciones indicadas en la Tabla 4.
|
Punto
de Medición |
Concentración
en Gas Patrón para Evaluar el Rango de Medición Inferior del SMCE |
Punto
de Medición |
Concentración
en Gas Patrón para Evaluar el Rango de Medición Superior del SMCE |
|
1 |
0-40
ppmv |
1 |
0-600
ppmv |
|
2 |
60-80
ppmv |
2 |
900-1200
ppmv |
|
3 |
140-160
ppmv |
3 |
2100-2400
ppmv |
Tabla
3.
Concentraciones de Gases
Patrón para la Prueba de EC
Registrar la concentración
reportada para cada gas en cada punto de medición. Repetir el procedimiento 3
veces de manera que se obtengan 3 respuestas del SMCE para cada punto y rango
de medición.
8.6 Procedimiento
de Prueba para la Evaluación del Tiempo de Respuesta (TR).
El Tiempo de Respuesta
aplica para todos los SMCE, sin embargo, generalmente es significativo sólo
para SMCE Extractivos.
8.6.1 Introduzca un
gas cero al SMCE por la sonda de la Interfase de Muestra. Espere a que la
lectura se estabilice de manera que no presente cambios mayores a un 1% del
LSMR durante 30 segundos. Cambie la alimentación del gas cero por la de un gas
de Nivel Medio o Alto y active un cronómetro. Registre el tiempo en que el SMCE
alcance un 95% de su valor de estabilización final en modo ascendente. Vuelva a
cambiar la alimentación a gas cero y registre el tiempo en que el SMCE alcanza
un 95% de su valor de estabilización final en modo descendente. Repita el
procedimiento anterior para lograr un total de 3 TR ascendentes y 3 TR
descendentes. Calcule el promedio para TR ascendente y descendente. El mayor de
ambos promedios de TR se considera el Tiempo de Respuesta del SMCE.
9.
Control de calidad
Ver inciso 13.
10.
Calibración y trazabilidad
Reservado.
11.
Procedimiento de análisis
No Aplica.
12.
Cálculos
12.1 Exactitud
Relativa.
Todos los datos de
concentración generados por el MR y SMCE deberán expresarse de manera
consistente en base seca y con las mismas unidades.
12.1.1 Corrección
por Humedad (cuando sea aplicable).
Realice una corrección a
Base Seca de aquellas concentraciones que se encuentren en Base Húmeda,
utilizando la siguiente ecuación:
![]()
Ecuación 8
Donde:
CBS = Concentración del analito en Base Seca.
CBH = Concentración del analito en Base Húmeda.
H = Fracción volumétrica
de vapor de agua en la muestra.
12.1.2 Corrección a
Condiciones Normales (cuando sea aplicable).
Cuando el Límite Máximo
Permisible de Emisión se exprese en Condiciones Normales, y las concentraciones
se tengan en unidades de partes por millón volumen, utilice la siguiente
relación para su corrección:
1 ppmv de CO = 1.144287 mg/m³ de CO a Condiciones
Normales (298ºK y 101325 Pa).
12.1.3 Corrección
por Dilución (cuando sea aplicable).
Cuando el Límite Máximo
Permisible de Emisión se exprese a una condición específica de dilución, y
cuando se haya realizado el monitoreo del diluyente correspondiente de manera
simultánea al MR y SMCE, realice la corrección conforme a la siguiente
ecuación:
Para O2 como
diluyente:
![]()
Ecuación 9
Donde:
CREF =
Concentración del analito Referida a un contenido de Oxígeno de Referencia.
C = Concentración del analito sin corrección por dilución.
%VO2,REF = Concentración de
Oxígeno de Referencia (% volumen, base seca).
El O2,REF en esta Norma es de 7 %v bs.
%VO2 = Concentración de Oxígeno Real (%
volumen, base seca).
12.1.4 Cálculo de la
Diferencia de Concentraciones entre MR y SMCE.
Calcule la diferencia de
concentraciones en cada corrida de MR y SMCE con la siguiente ecuación:
![]()
Ecuación 10
Donde:
Di = Diferencia entre la Concentración Promedio
obtenida por MR y SMCE durante la muestra o corrida número “i”.
CMR,i= Concentración Promedio del analito obtenida
por MR en la muestra o corrida número “i”.
CSMCE,i = Concentración Promedio del analito obtenida
por SMCE en la muestra o corrida número “i”.
12.1.5 Cálculo de la
Diferencia Promedio de Concentraciones entre MR y SMCE.
Calcule la diferencia de
concentraciones promedio en cada corrida de MR y SMCE con la siguiente
ecuación:

Ecuación 11
Donde:
DPROM =
Diferencia Promedio entre las Concentraciones Promedio obtenidas por MR y SMCE
durante todas las corridas o muestras.
Di = Diferencia
entre la Concentración Promedio obtenida por MR y SMCE durante la muestra o
corrida número “i”.
M = Número total de
corridas o muestras tomadas para la prueba de ER.
12.1.6 Cálculo de
Desviación Estándar de dPROM.
Calcule la desviación
estándar de dPROM con la siguiente ecuación:

Ecuación 12
Donde:
Sd = Desviación
Estándar de dPROM.
Di = Diferencia
entre la Concentración Promedio obtenida por MR y SMCE durante la muestra o
corrida número “i”.
M = Número total de
corridas o muestras tomadas para la prueba de ER.
12.1.7 Coeficiente
de Confianza (CC).
Calcule el Coeficiente de
Confianza usando la siguiente ecuación:
![]()
Ecuación 13
Donde:
CC = Coeficiente de Confianza.
Sd = Desviación
Estándar de dPROM.
t0.975 = Valor de Distribución t Student a 97.5% de Confianza (Ver Tabla 4).
M = Número total de
corridas o muestras tomadas para la prueba de ER.
|
M |
t0.975 |
M |
t0.975 |
M |
t0.975 |
|
2 |
12.706 |
7 |
2.447 |
12 |
2.201 |
|
3 |
4.303 |
8 |
2.365 |
13 |
2.179 |
|
4 |
3.182 |
9 |
2.306 |
14 |
2.160 |
|
5 |
2.776 |
10 |
2.262 |
15 |
2.145 |
|
6 |
2.571 |
11 |
2.228 |
16 |
2.131 |
Tabla
4.
Valor de distribución t
student a M-1 grados de libertado y 97.5%
de confianza
12.1.8 Cálculo de la
Exactitud Relativa (ER).
Calcule ER con la siguiente ecuación:
![]()
Ecuación 14
Donde:
ER = Exactitud Relativa
(%).
CC = Coeficiente de
Confianza.
DPROM =
Diferencia Promedio entre las Concentraciones Promedio obtenidas por MR y
SMCE durante todas las corridas o
muestras.
CMR,PROM =
Concentración Promedio del analito obtenida en todas las corridas o muestras
por MR.
12.2 Error de
Calibración (EC).
Con los datos registrados
en la prueba del inciso 8.5, calcule
el Error de Calibración con la siguiente ecuación:
![]()
Ecuación 15
Donde:
ECi,j =Error de Calibración para el Punto de
Medición “i” en el Rango de Medición “j” (ver Tabla 3), (%).
CSMCE,PROM,i,j=Concentración
Promedio del SMCE ante el Gas Patrón utilizado para el Punto de Medición “i” en
el Rango de Medición “j”
(ver Tabla 3).
CPATRON,i,j = Concentración del
Gas Patrón utilizado para el Punto de Medición “i” en el Rango de Medición “j”
(ver Tabla 3).
LSMR = Límite Superior de
Medición o Registro para el Rango de Medición “j”.
Nota: se deberá ser
congruente con las unidades de C y LSMR.
Nota: en la prueba
de EC, el término “Punto de Medición” se refiere a “Nivel de Concentración”.
13.
Especificaciones para pruebas de desempeño
13.1 Sistema de
Adquisición de Datos o Registro.
Deberá cumplir con las
indicaciones del inciso 6.1.1 y
Tabla 1.
13.2
Desplazamiento de Calibración.
El SMCE debe permitir la
determinación del Desplazamiento de Calibración en los valores cero y nivel
alto. La respuesta del SMCE a la calibración no debe desviarse del valor de
referencia del gas patrón, celdas de gas patrón o filtros ópticos, por más del
± 3% del LSMR por cada 24 horas para la prueba de 7 días.
13.3 Exactitud
Relativa.
Los resultados de la prueba
de ER para el SMCE de CO no deben ser mayores al 10% cuando se utilice el valor
promedio obtenido por el MR como denominador en la Ecuación 12, o 5% cuando se
utilice el Límite Máximo Permisible de Emisión asignado al ducto como
denominador en la Ecuación 12 (en substitución de CMR,PROM), o estar
dentro de 5 ppmv de CO cuando se calcule ER como diferencia absoluta promedio
más el coeficiente de confianza CC.
13.4 Tiempo de
Respuesta.
El Tiempo de Respuesta no
deberá estar ser mayor a 2 minutos.
13.5 Error de
Calibración.
El Error de Calibración
para cada Punto de Medición y Rango de Medición (ver Tabla 3) no debe ser mayor
al ± 5% del LSMR.
14.
Procedimiento alterno para la prueba de exactitud relativa
Bajo ciertas condiciones de
operación es posible que no se obtengan resultados significativos con el
Procedimiento de Prueba de Exactitud Relativa (ER). Esto incluye aquellas
condiciones en que la concentración de CO se encuentre de manera consistente en
niveles bajos o con interrupciones periódicas de alta concentración de corta
duración. En estas situaciones, es apropiada la substitución de la prueba de ER
por el siguiente procedimiento.
Conduzca una verificación
completa del sistema de monitoreo de acuerdo a las especificaciones del
fabricante. Esta verificación debe incluir: (1) operación de la fuente de luz;
(2) receptor de la señal; (3) programación de funciones; (4) sistema de
adquisición de datos; (5) funciones de reducción de datos; (6) registradores;
(7) funciones operadas mecánicamente; (8) filtro de muestras; (9) líneas de
calentamiento de muestra (cuando sea aplicable); (10) trampas de humedad, y
cualquier otra función aplicable al SMCE. Todas las partes del SMCE deberán de
funcionar apropiadamente antes de que la prueba de Exactitud Relativa pueda ser
omitida. El instrumento deberá aprobar las pruebas de Error de Calibración (EC)
y Desplazamiento de Calibración (DC). Cualquier substitución del procedimiento
alterno deberá ser autorizada.
15.
Ejemplos de pruebas de desempeño
A continuación se presentan
ejemplos numéricos para cada prueba de desempeño.
15.1
Desplazamiento de Calibración.
|
Día de Prueba “i” |
Fecha (dd/mm/aa) |
Rango de Medición |
LMSR (ppmv) |
Nivel de Conc. |
Concentración en Gas Patrón (ppmv) |
Concentración de Respuesta Estable del SMCE (ppmv) |
Desplazamiento de Calibr. Ci (%) |
Espec. Máxima para DCi (%) |
|
1 |
1/01/01 |
Inferior |
200 |
Cero |
0 |
5 |
2.5 |
3 |
|
|
|
Superior |
3000 |
Alto |
2400 |
2463 |
2.1 |
3 |
|
2 |
2/01/01 |
Inferior |
200 |
Cero |
0 |
1 |
0.5 |
3 |
|
|
|
Superior |
3000 |
Alto |
2400 |
2415 |
0.5 |
3 |
|
3 |
3/01/01 |
Inferior |
200 |
Cero |
0 |
0 |
0.0 |
3 |
|
|
|
Superior |
3000 |
Alto |
2400 |
2379 |
0.7 |
3 |
|
4 |
4/01/01 |
Inferior |
200 |
Cero |
0 |
2 |
1.0 |
3 |
|
|
|
Superior |
3000 |
Alto |
2400 |
2460 |
2.0 |
3 |
|
5 |
5/01/01 |
Inferior |
200 |
Cero |
0 |
2 |
1.0 |
3 |
|
|
|
Superior |
3000 |
Alto |
2400 |
2411 |
0.4 |
3 |
|
6 |
6/01/01 |
Inferior |
200 |
Cero |
0 |
2 |
1.0 |
3 |
|
|
|
Superior |
3000 |
Alto |
2400 |
2389 |
0.4 |
3 |
|
7 |
7/01/01 |
Inferior |
200 |
Cero |
0 |
5 |
2.5 |
3 |
|
|
|
Superior |
3000 |
Alto |
2400 |
2388 |
0.4 |
3 |
15.2 Error de
Calibración.
|
Prueba |
Rango
de Medición "j" |
Punto
de Medición "i" |
LMSR
(ppmv) |
Concentración
en Gas Patrón (ppmv) |
Concentración
de Respuesta Estable del SMCE (ppmv) |
Error
de Calibración ECi,j (%) |
Especificación
Máxima para ECi,j (%) |
|
|
|
1 |
200 |
0 |
5 |
2.5 |
5 |
|
1 |
Inferior |
2 |
200 |
70 |
69 |
0.5 |
5 |
|
|
|
3 |
200 |
150 |
152 |
1.0 |
5 |
|
|
|
1 |
3000 |
200 |
215 |
0.5 |
5 |
|
1 |
Superior |
2 |
3000 |
1000 |
903 |
3.2 |
5 |
|
|
|
3 |
3000 |
2200 |
2145 |
1.8 |
5 |
|
|
|
1 |
200 |
0 |
2 |
1.0 |
5 |
|
2 |
Inferior |
2 |
200 |
70 |
76 |
3.0 |
5 |
|
|
|
3 |
200 |
150 |
142 |
4.0 |
5 |
|
|
|
1 |
3000 |
200 |
215 |
0.5 |
5 |
|
2 |
Superior |
2 |
3000 |
1000 |
1100 |
3.3 |
5 |
|
|
|
3 |
3000 |
2200 |
2220 |
0.7 |
5 |
|
|
|
1 |
200 |
0 |
0 |
0.0 |
5 |
|
3 |
Inferior |
2 |
200 |
70 |
75 |
2.5 |
5 |
|
|
|
3 |
200 |
150 |
153 |
1.5 |
5 |
|
|
|
1 |
3000 |
200 |
198 |
0.1 |
5 |
|
3 |
Superior |
2 |
3000 |
1000 |
1050 |
1.7 |
5 |
|
|
|
3 |
3000 |
2200 |
2280 |
2.7 |
5 |
Obteniéndose los siguientes
errores promedio para cada punto y rango de medición:
EC1,INFERIOR
= 1.17
EC2,INFERIOR
= 2.00
EC3,INFERIOR
= 2.17
EC1,SUPERIOR
= 0.36
EC1,SUPERIOR
= 2.74
EC1,SUPERIOR
= 1.72
Siendo todos menores al
criterio de aceptación de ±5%.
15.3 Exactitud
Relativa.
|
Número
de Corrida de MR |
Fecha (dd/mm/aa) |
Inicio (hh:mm) |
Término (hh:mm) |
Concentración
Promedio Obtenida por MR (ppmv) |
Concentración
Promedio Obtenida por SMCE (ppmv) |
Diferencia
de Concentraciones di (ppmv) |
|
1 |
1/01/01 |
8:23 |
9:23 |
52 |
48 |
4 |
|
2 |
1/01/01 |
11:45 |
12.45 |
963 |
993 |
-30 |
|
3 |
1/01/01 |
15:18 |
16:18 |
103 |
110 |
-7 |
|
4 |
1/01/01 |
17:30 |
18:30 |
52 |
58 |
-6 |
|
5 |
2/01/01 |
9:03 |
10:03 |
22 |
33 |
-11 |
|
6 |
2/01/01 |
12:08 |
13:08 |
83 |
96 |
-13 |
|
7 |
2/01/01 |
15:00 |
16:00 |
45 |
42 |
3 |
|
8 |
4/01/01 |
12:15 |
13:15 |
23 |
21 |
2 |
|
9 |
4/01/01 |
14:25 |
15:25 |
15 |
14 |
1 |
Obteniéndose la siguiente
Exactitud Relativa:
CMR,PROM
= 150.89 ppmv
M =
9
DPROM
= -6 ppmv
Sd= 10.86 ppmv
T = 2.306
CC = 8.35
ER = 9.73%
Siendo el ER obtenido menor
al criterio de aceptación de ±10% del MR.
15.4 Tiempo de
Respuesta.
|
Tipo de Prueba |
Número de Prueba |
Tiempo de Respuesta (s) |
|
|
1 |
123 |
|
Ascendente |
2 |
145 |
|
|
3 |
132 |
|
|
1 |
187 |
|
Descendente |
2 |
188 |
|
|
3 |
173 |
Resultando en los
siguientes promedios:
TRASCENDENTE
= 133 s
TRDESCENDENTE
= 183 s
De manera que el Tiempo de
Respuesta del SMCE es de:
TR = 183 s
Dado que el criterio de
aceptación para TR es de 2 minutos (120 segundos), el SMCE tendría que
modificarse para cumplir con este criterio.
16. Bibliografía
16.1 Jahnke, James A. And G.J. Aldina “Handbook
Continuous Air Pollution source monitoring systems”. U.S. Environmental Protection Agency Technology Transfer
Cincinnati, Ohio 45268 EPA-625/6 79/005.
16.2 Michie Raymand, M Jret
al., Performance Test and comparative data for designated Reference method for
Carbon monoxide U.S.- Environmental Protection
Agency ORD/EMLS, EPA 600/S4-83-013 September 1982.
ANEXO 2
DETERMINACIÓN
DE EMISIONES DE ÓXIDOS DE NITRÓGENO EN
FUENTES FIJAS
(PROCEDIMIENTO DE ANÁLISIS INSTRUMENTAL)
ÍNDICE
1. Objetivo y principio
2. Intervalo de medición y sensibilidad
3. Definiciones
4. Especificaciones de desempeño para el
sistema de medición
5. Equipamiento y reactivos
6. Procedimiento de prueba para el
desempeño del sistema de medición
7. Procedimiento de pruebas de
emisión
8. Cálculos de emisión
9. Bibliografía
1.
Objetivo y principio
1.1 Objetivo
Este método establece el
procedimiento para la determinación de concentraciones de emisión de óxidos de
nitrógeno (NOx), provenientes de fuentes fijas.
1.2 Principio
Una muestra se extrae
continuamente de una corriente de gases que fluyen por conducto, dicha muestra
es llevada a un analizador que opera por el principio de quimiluminiscencia
para la determinación de las concentraciones de NOx.
2. Intervalo de
medición y sensibilidad
2.1 Intervalo
analítico
El intervalo analítico es
determinado por el diseño del instrumento. Para este método el intervalo
analítico deberá ser ajustado por la elección del LIMITE SUPERIOR DE MEDICION
DE REFERENCIA (LSMR) del sistema de monitoreo. El LMSR para el sistema de
monitoreo debe seleccionarse de tal manera que la concentración del gas
contaminante sea equivalente al estándar de emisión, éste no deberá ser menor
al 30 por ciento del LMSR. Si durante el periodo de prueba de la corrida, la
concentración del gas contaminante excediera la concentración del LMSR, la
corrida será considerada inválida.
2.2 Sensibilidad
La sensibilidad depende del
intervalo analítico (rango) depende del intervalo analítico, el LSMR y el ruido
en la señal del sistema de medición. Para un buen diseño del sistema, el límite
mínimo detectable deberá ser menor al 2 por ciento del LMSR.
3.
Definiciones
Sistema de medición.-
Consiste de los siguientes subsistemas:
3.1 Respuestas
por interferencias
La respuesta generada por
el sistema de medición para un componente del gas muestreado, que no
corresponde al componente del gas que está siendo medido.
3.1.1 Interfase de
muestra
La porción de un sistema
usado por uno o más de los siguientes componentes: toma de muestra, transporte
de muestra, acondicionamiento de muestra o accesorios para protección de los
analizadores de los posibles efectos que el efluente presente en la emisión de
la fuente.
3.1.2 Analizador de
gas
La porción del sistema que
se activa por el paso del gas en el analizador y que genera una respuesta
proporcional para esa concentración.
3.1.3 Registro de
datos
Un registrador gráfico, un
sistema de adquisición de datos o registrador digital para anotar los datos
medidos por el analizador.
3.1.4 Convertidor
de NO2 a NO
Un dispositivo que
convierte el dióxido de nitrógeno (NO2) presente en la muestra de
gas a óxido de nitrógeno (NO).
3.2 LSMR
Límite superior de
concentración de cada intervalo de medición del sistema de adquisición de
datos, registrador y/o analizador de gases
3.3 Gas de
calibración
Una concentración conocida
del gas de interés en una mezcla diluida con un gas inerte.
3.4 Error de
calibración del analizador
La diferencia entre la
concentración de gas exhibida por el analizador del gas y la concentración
conocida del gas de calibración, cuando el gas de calibración es introducido
directamente al analizador.
3.5 Desviación
interfase de muestra (DIM)
Sesgos del sistema de
muestreo
La diferencia entre las
concentraciones del gas exhibidas por el sistema de medición cuando una
concentración conocida del gas es introducida a la salida de la sonda de
muestreo y cuando el mismo gas es introducido directamente al analizador.
3.6 Cero de
calibración
La diferencia entre la
lectura generada por el sistema de medición por la respuesta inicial de
calibración al nivel de concentración cero, luego del periodo establecido de
operación durante el cual no haya ocurrido operaciones de mantenimiento,
reparación o ajuste, no programado.
3.7
Desplazamiento de calibración
La diferencia entre la
lectura generada por el sistema de medición por la respuesta inicial de
calibración al nivel de concentración en el intervalo mínimo, luego del periodo
establecido de operación durante el cual no haya ocurrido operaciones de
mantenimiento, reparación o ajuste, no programado.
3.8 Tiempo de
respuesta
Periodo de tiempo requerido
por el sistema de medición para desplegar el 95 por ciento de un cambio en la
concentración del gas en el registrador de datos.
4.
Especificaciones de desempeño para el sistema de medición
4.1 Error de
calibración del analizador
Valores menores que el ± 2
por ciento del LSMR para los valores del cero analítico, del nivel mínimo y
máximo para los gases de calibración.
4.1.1 Desviación de
la interfase de la muestra (DIM)
Valores menores que el ± 5
por ciento del LSMR, para valores del cero analítico, del nivel mínimo de los
gases de calibración.
4.1.2 Ajuste del
cero
Valor menor que ± 3 por
ciento del LSMR sobre el periodo de cada corrida
4.1.3 Ajuste de
calibración
Valores menores que ± 3 por
ciento del intervalo de determinación sobre el periodo de cada corrida.
5.
Equipamiento y reactivos
5.1 Sistema de
medición
Utilice cualquier sistema
de medición para NOx que satisfaga las especificaciones de este método. Un
diagrama de un sistema aceptable de medición se muestra en la Figura 1. Los componentes esenciales del
sistema de medición se describen a continuación:
5.1.1 Sonda de
muestreo
Sonda fabricada en vidrio,
acero inoxidable o equivalente, de suficiente longitud para alcanzar los puntos
transversales de muestreo. A la sonda de muestreo preferentemente debe dársele
calentamiento para evitar la condensación interna de los gases muestreados.
5.1.2 Línea de
muestreo
Constituida de acero
inoxidable con temperatura suficiente (caliente para evitar la condensación), o
tubo de PTFE, para transportar la muestra de gas hasta el sistema de remoción
de humedad.
5.1.3 Líneas de
transporte de muestra
Tuberías de acero
inoxidable o de PTFE, para transportar la muestra desde el sistema de remoción
de humedad hasta la bomba de muestreo, el controlador de flujo de muestra y la
válvula selectora para la muestra gaseosa.
5.1.4 Ensamble de
válvula de calibración
Una válvula con ensamble de
tres vías o equivalente para conducir el flujo de gas de la muestra e introducción
de los gases de calibración al sistema de medición directamente en la salida de
la sonda de muestreo cuando está en el modo de calibración.
5.1.5 Sistema de
remoción de humedad
Un condensador tipo
serpentín frío o equipo similar (esto es un secado permeable), para remover
continuamente el líquido condensado a partir de la muestra de gas mientras se
tenga un contacto mínimo entre el condensador y la muestra de gas. El sistema
de remoción de humedad no es necesario para analizadores que puedan medir la
concentración de gas en base húmeda. Para estos analizadores efectúe las
siguientes operaciones: (1) Caliente la línea de muestreo y todo los
componentes de la interfase lo suficiente para prevenir condensación a la
entrada del analizador y (2) determine el contenido de humedad y corrija la
lectura de la medición de la concentración del gas en base seca usando los
métodos apropiados, sujeto a la aprobación del técnico responsable del
muestreo. La determinación del contenido de humedad en la muestra no es
necesaria para analizadores de contaminantes que midan su concentración en base
húmeda cuando: (1) el analizador de CO2 en base húmeda es usado para
obtener mediciones simultáneas y (2) las mediciones se reportan como la
relación entre concentraciones de contaminantes /CO2 usadas para
determinar las emisiones equivalentes medidas o los límites máximos de emisión
normales.
5.1.6 Filtro
colector de partículas
El filtro colector de
partículas puede estar instalado dentro de la chimenea o afuera de la misma,
teniendo la precaución de calentar lo suficiente para evitar el agua de
condensación, dentro de la línea de muestreo. El filtro deberá ser de lana de
vidrio de borosilicato, cuarzo o materiales de fibra de vidrio. Pueden ser
utilizados filtros adicionales a la entrada o a la salida del sistema de
remoción de humedad y a la entrada del analizador para prevenir la acumulación
de partículas sobre el sistema de medición y prolongar la vida útil de los
componentes, todos los filtros habrán de ser fabricados con material inerte al
gas a ser muestreado.
5.1.7 Bomba de
muestreo
Una bomba hermética para
extraer la muestra de gas a través del sistema a un caudal suficiente de flujo
para minimizar el tiempo de respuesta del sistema de medición. La bomba puede
ser construida por cualquier material que no reaccione con el gas a muestrear.
5.1.8 Control de
flujo de la muestra
Una válvula controladora
del caudal de flujo de la muestra y un rotámetro o equivalente para mantener un
caudal de muestreo constante dentro de un 10 por ciento. (Nota: El técnico
responsable de las pruebas puede instalar un regulador de presión a la válvula
de muestreo del gas, para mantener una presión constante de tal manera que se
proteja a los analizadores de sufrir una presurización excesiva y reducir las
necesidades en el ajuste a los flujos manejados por el sistema).
5.1.9 Válvula
selectora para la muestra gaseosa
Válvula selectora para la
muestra gaseosa, para desviar una porción de la corriente de la muestra gaseosa
al analizador y al resto hacia una válvula de venteo de descarga. La válvula
selectora para la muestra gaseosa también puede incluir provisiones para la
introducción de gases de calibración directamente al analizador. La válvula
selectora puede ser construida con cualquier material que no reaccione con el
gas a muestrear.
5.1.10 Registrador
de datos
Registrador gráfico de papel, sistema de adquisición de datos o registro digital, para anotar los datos medidos. La resolución del registrador de datos debe ser de 0.5 por ciento del intervalo de determinación. Alternativamente, un medidor digital o analógico da una resolución de 0.5 por ciento del intervalo de determinación, puede ser usado para obtener las respuestas del analizador y las lecturas pueden ser anotadas manualmente. Si estas alternativas son usadas, las lecturas pueden ser obtenidas a espacios de intervalos iguales, sobre la duración de la corrida de muestreo debe ser menos a una hora en intervalos de medición de 1 minuto o un mínimo de 30 mediciones, cualquiera que sea menos restrictiva. La duración de las corridas de muestreo mayores a 1 hora, se debe medir en intervalos de 2 minutos o mínimo de 96 mediciones, cualquiera que sea menos restrictiva.
5.1.11 Convertidor
de NO2 a NO
Es la parte del sistema que
convierte el NO2 presente en el gas muestreado a NO; un convertidor
del NO2 a NO no es necesario si la cantidad de NO2
presente en el gas de escape es menor al 5 por ciento de la concentración total
de NOx.
5.1.12 Analizador de
NOx
Un analizador basado en los
principios de quimiluminiscencia puede determinar continuamente las
concentraciones de NOx en la corriente de gas muestreada. El analizador debe
satisfacer las especificaciones aplicables al desempeño descritas en la sección
4. Se habrá de adaptar al analizador
un contador de caudal (esto es, un rotámetro de precisión y un barómetro de
presión, anexo a todos los controladores del caudal).
5.2 Gases de
calibración de NOx
Los gases de calibración de
NOx para el analizador de NOx deberán ser mezclas de NO balance N2
use cuatro mezclas de gases de calibración:
Dos concentraciones de
gases de calibración, cero y una concentración equivalente de 80 hasta 100 por
ciento del intervalo de medición. El gas de calibración deberá tener una
concentración mayor a la concentración de los gases a medir. El aire
prepurificado puede ser utilizado para calibrar el blanco analítico pasando el
aire a través de un filtro de carbón activado a través de uno o más
burbujeadores que contengan una solución acuosa al 3 por ciento de peróxido de
hidrógeno (H2O2).
5.2.1 Use aire
ambiente como gas de calibración para el cero analítico.
6. Procedimiento
de prueba para el desempeño del sistema de medición
Llevar a cabo los
siguientes procedimientos antes de realizar las mediciones a las emisiones
(sección 7).
6.1 Verificación de las
concentraciones de los gases de calibración
6.1.1 Uso de gases
de calibración que son certificados por el fabricante de mezclas de gases que
satisfagan el protocolo.
6.2 Preparación
del sistema de medición
Ensamble el sistema de
medición siguiendo el manual de instrucciones del fabricante, preparación y preacondicionamiento del analizador de gas
y de ser posible; a los otros componentes del sistema. Introduzca los gases de
calibración en cualquier secuencia y realice todos los ajustes necesarios para
calibrar el analizador y el registrador de datos. Ajuste los componentes del
sistema para obtener el caudal correcto de muestreo.
6.3 Error de
calibración del analizador
Realice la revisión del
error de calibración en el analizador introduciendo gases de calibración al
sistema de medición en cualquier punto corriente arriba del analizador de gases
como sigue:
6.3.1 Luego que el
sistema de medición halla sido preparado para su uso introduzca los gases de
calibración para el cero analítico y valor máximo de la curva de calibración al
analizador. Durante esta revisión no realice ajuste al sistema excepto aquellos
que consigan el caudal de flujo correcto de los gases de calibración al
analizador. Anote la respuesta del analizador para el gas de calibración dentro
del Formato 6-1. (Nota: La curva de calibración
establecida previamente al paso de revisión del error de calibración del
analizador puede usarse para convertir la respuesta del analizador a la
concentración equivalente del gas introducido al analizador. Sin embargo, el
mismo procedimiento de corrección debe usarse para todas las mediciones de los
gases de calibración y de los efluentes obtenidos durante la corrida).
6.3.2 La revisión
del error de calibración del analizador debe considerarse inválida si la
concentración del gas exhibida por el analizador excede ± 2 por ciento del
intervalo de medición para cualquiera de los gases de calibración. Si se
excediera una calibración de intervalo inválida, lleve a cabo acciones
correctivas y repita la revisión en el error de calibración del analizador
hasta lograr el desempeño aceptable.
6.4 Revisión del
DIM por el sistema de muestreo
Revise el sesgo causado por
el sistema de muestreo de introducción de gases de calibración instalada en la
salida de la sonda de muestreo. Un gas de calibración para el blanco analítico
y ya sea un gas de calibración del intervalo mínimo o de valor máximo de la
curva de calibración aquel que se aproxime más a las concentraciones del contenido
en el efluente deberá ser usado para su revisión como sigue:
6.4.1 Introduzca el
gas de calibración para la curva de calibración y anote la concentración del
gas que exhibe el analizador dentro del Formato 6-1. Luego introduzca el gas de calibración del blanco analítico y
anote la concentración de gas exhibida por el analizador o durante la revisión
del DIM por el sistema de muestreo opere el sistema al caudal de flujo normal
de muestreo y no realice ajuste a los sistemas de medición exceptuando aquellos
que sean necesarios para lograr los caudales de flujo apropiados de los gases
de calibración e introducidos al analizador. Alternadamente introduzca los
gases de calibración para el cero analítico y para la curva de calibración
hasta lograr una respuesta estable. El técnico responsable deberá determinar el
tiempo para el sistema de medición observando los tiempos requeridos para
lograr la respuesta estable para ambos gases de calibración para el blanco
analítico y para la curva de calibración. Anotar cual de los dos tiempos es
mayor y considere como tiempo de respuesta.
6.4.2 La revisión
del DIM para el sistema de muestreo debe considerarse inválido si la diferencia
entre la concentración de los gases exhibidas por el sistema de muestreo
determinada durante la revisión de error de calibración del analizador y la
revisión del DIM para el sistema de muestreo excede ± 5 por ciento de la escala
de la curva de calibración. Si se exhibe cualquier calibración inválida, lleve
a cabo acciones correctivas y repita la revisión del error en la calibración
del analizador y luego repita la revisión del DIM para el sistema de muestreo.
7.
Procedimiento de prueba de emisiones
7.1 Selección del
sitio de muestreo y punto de muestreo
El punto de muestreo deberá
ser en el centro de la sección transversal de la fuente a la altura de los
puertos.
7.2 Colección de
la muestra
Coloque la sonda de
muestreo en el punto de medición, e inicie el muestreo al mismo caudal que se
usó durante las pruebas del tiempo de respuesta, mantenga constante el caudal
de muestreo (esto es, dentro del ±10 por ciento) a lo largo de toda la corrida;
el tiempo de muestreo por corrida debe ser el mismo que el tiempo total
requerido para llevar a cabo una corrida adicionando el doble del tiempo promedio
de respuesta al sistema.
Para la corrida use sólo mediciones obtenidas luego de haber transcurrido el
doble de tiempo de respuesta del sistema de medición para determinar la
concentración promedio del contenido en el efluente.
7.3 Pruebas de cambio en el intervalo de
calibración y el cero analítico
Inmediatamente antes y
después de cada corrida o para cuando sea necesario el ajuste al sistema de
medición, durante la corrida repita el procedimiento de revisión del DIM para
el sistema de muestreo descrito en la sección 6.5 (No realice ajuste al sistema de medición hasta después que se
hallan realizado completamente las revisiones en las tendencias). Anote la
respuesta del analizador dentro del formato 6-3.
7.3.1 Ya sea que
los valores de calibración para el blanco analítico o para la curva de
calibración excedan lo especificado para el DIM para el sistema de muestreo,
entonces considere inválida la corrida. Repita ambos procedimientos de revisión
de error en la calibración del analizador (sección 6.4) y procedimiento de revisión del DIM para sistema de muestreo
(sección 6.5) antes de cada corrida.
7.3.2 En caso de
valores de calibración para el cero analítico y para la curva de calibración
del DIM para el sistema de muestreo, use el promedio de los valores inicial y
final de la revisión del DIM para calcular la concentración de gas para la
corrida. Si el valor en la tendencia para la calibración del cero analítico o
para la curva de calibración excede los límites de tendencia basados en la
diferencia entre las respuestas de revisión del DIM para sistemas de muestreo
inmediatamente antes y después de la corrida, repita el procedimiento de
revisión del DIM para el sistema de muestreo (sección 6.5) antes de realizar corridas adicionales.
8.
Cálculos de emisión
La concentración media del
gas en el efluente se calcula a partir de concentraciones medias del gas
exhibidas para el analizador de gas y es ajustado por las revisiones del DIM
para el sistema de muestreo para el cero analítico y para la curva de calibración,
como se determina en la sección 7.3.
La concentración media del gas exhibida por el analizador puede calcularse
mediante la integración del área bajo la curva usando registradores de
impresión en papel, o al promediar todas las mediciones válidas del efluente.
De manera alternativa el promedio puede calcularse de las mediciones anotadas
homogéneamente a lo largo de todo el tiempo que duró la corrida. Para muestras
de tiempos de corrida menores a 1 hora, las mediciones a intervalos de 1 minuto
o con un mínimo de 30 mediciones cualquiera que sea el menor registrado debe
utilizarse. Para el tiempo de duración del muestreo para la corridas, máximas a
1 hora deben usarse las mediciones a intervalos de 2 minutos o con un mínimo de
96 mediciones. Calcule la concentración del gas en el efluente usando la
siguiente ecuación:
Ecuación:
Cgas = (Cprom - Co) Cmax./
Cpae - Co
Donde:
Cgas= Concentración del gas
en el efluente, en base seca, ppm.
Cprom = Concentración
promedio del gas indicada por el analizador del gas, en base seca, ppm.
Co=Promedio máximo de los
valores inicial y final durante la revisión del DIM del sistema de calibración
alimentado con el gas del cero analítico, en ppm.
Cpae =Promedio de los
valores de la revisión inicial y final del DIM del sistema de calibración para
el gas alimentado en la calibración de la curva de calibración, ppm.
Cmax = Concentración máxima del gas de calibración analizado para la
curva de calibración, ppm.
9.
Bibliografía
9.1 Protocolo de
Trazabilidad para el Establecimiento de concentraciones Exactas para Gases
Usados para Calibración y Auditorías de Monitores de Emisión Fuentes Continuas:
Protocolo Número 1. Agencia de
Protección Ambiental, E.U.A., División de Aseguramiento de la Calidad. Parque
de Investigación Triángulo. N.C. Junio de 1978.
9.2 Westin, Peter
R. y John Brown, Métodos para Colección y Análisis de Muestras de Gas en
Cilindros. Sociedad de Evaluación de Fuentes Newsletter. 3 (3) . 5-15. Septiembre
de 1978.
9.3 Emission Measurement
Technical Information Center NSPS Test Method. Method 6C-Determination of
Sulphur Dioxide Emissions from Stationary Sources.
FORMATO
6-1 Análisis de Datos de Calibración
|
Fuente de Identificación: |
Corrida: |
|||||
|
Técnico: |
Intervalo de
determinación |
|||||
|
Dato: |
||||||
|
Datos de Calibración del
Analizador para el Muestreo |
||||||
|
|
Valor del Cilindro
(indicando Unidades) |
Respuesta del Analizador
de Calibración (indicando Unidades) |
Diferencia Absoluta
(indicando Unidades) |
Diferencia (porcentaje de intervalo
de determinación) |
||
|
Gas Cero |
|
|
|
|
||
|
Gas de Intervalo mínimo
en la curva de calibración |
|
|
|
|
||
|
Gas de Intervalo Máximo
en la curva de calibración |
|
|
|
|
||
FORMATO
6-2 Sistema de DIM Calibración y Tendencia de Datos
|
Fuente de Identificación: |
Número de Corrida: |
|||||||
|
Técnico: |
Intervalo de
determinación |
|||||||
|
Dato: |
||||||||
|
|
|
Valores Iniciales |
Valores Finales |
|||||
|
|
Respuesta del analizador
de calibración |
Respuesta del sistema de
calibración |
Sistema de DIM
Calibración (% intervalo de
determinación) |
Sistema de DIM
Calibración (% intervalo de
determinación) |
Respuesta del sistema de calibración |
Tendencia (% intervalo de
determinación) |
||
|
Gas cero |
||||||||
|
Alta escala de s |
||||||||
![]()
![]()
ANEXO 3
MÉTODO
DE ANÁLISIS PARA DETERMINACIÓN DE METALES EN EMISIONES
DE
FUENTES FIJAS POR ESPECTROFOTOMETRÍA DE ABSORCIÓN ATÓMICA
ÍNDICE
0. Introducción
1. Objetivo y
campo de aplicación
2. Referencias
3.
Interferencias
4. Definiciones
5. Equipo y
materiales
6. Reactivos y
patrones
7. Recolección,
preservación y almacenamiento de muestras
8. Control de
calidad
9. Calibración
10. Procedimiento
11. Cálculos
12. Seguridad
13. Desempeño del
método
14. Manejo de
residuos
15. Bibliografía
16. Tablas y
figuras
0.
Introducción
Este método se basa en qué
muestras provenientes de emisiones de fuentes fijas son sometidas a una
digestión ácida para su posterior análisis de metales por la técnica de
espectrofotometría de absorción atómica, se fundamenta en que cuando un haz de
energía radiante monocromática incide sobre una muestra atomizada, parte de la
energía es absorbida y el resto transmitida, la cantidad de luz absorbida por
el elemento atomizado en una flama es medida por un detector, si la energía
radiante incidente tiene longitudes de onda de la región visible del espectro y
del medio a través del que tiene que pasar, absorbe selectivamente ciertas
longitudes de onda, por lo tanto la cantidad de energía absorbida en la flama a
una cierta longitud de onda es proporcional a la concentración del analito de
interés.
1.
Objetivo y campo de aplicación
1.1 Objetivo
Establecer un método de
prueba para la determinación de metales en emisiones de fuentes fijas por
Espectrofotometría de Absorción Atómica.
1.2 Campo de
aplicación
Este método aplica para la
determinación de los siguientes metales Arsénico (As), Selenio (Se), Cobalto
(Co), Níquel (Ni), Manganeso (Mn), Estaño (Sn), Cadmio (Cd), Plomo (Pb), Cromo
total (Cr Tot.), Cobre (Cu), Zinc (Zn), Mercurio (Hg) en emisiones
de fuentes fijas.
2. Referencias
Este
método se complementa con la siguiente norma mexicana vigente:
2.1
NOM-008-SECOFI-1993.- Sistema
general de unidades de medida.
3.
Interferencias
3.1
Interferencias para espectrofotometría de absorción atómica con aditamento de
vapor frío. Debe utilizarse agua desionizada durante el muestreo y la
preparación de estándares. Cualquier compuesto con la misma absorbancia en la
longitud de onda del metal de interés puede causar una interferencia positiva.
Algunos compuestos orgánicos volátiles (p.e., benceno, tolueno, acetona,
tetracloruro de carbono) absorben a la longitud de onda en que se lee el
mercurio y son consideradas interferencias analíticas. Esto ocurre como
contaminación en los reactivos usados durante la preparación de muestra.
3.2 El incremento
de la concentración del ácido nítrico en las muestras o estándares produce una
elevada señal de fondo. La concentración del ácido nítrico en las muestras y
estándares no debe ser mayor de 10%.
4.
Definiciones
4.1 Emisión.-
Descarga a la atmósfera de toda sustancia en cualquiera de sus estados físicos
o de energía.
4.2 PTFE.- Nombre
común que se le da a la sustancia química politetrafluoroetileno, se usa
generalmente para cubrir o proteger superficies en forma inerte.
5.
Equipo y materiales
Sólo se mencionan los
equipos y materiales que son de relevancia para el presente método.
5.1 Equipo para
el análisis
5.1.1 Balanza analítica con precisión de 0,1 mg
5.1.2 Espectrofotómetro
de Absorción Atómica (EAA): Con haz sencillo o doble, monocromador, detector,
fotomultiplicador ajustable al ancho de banda espectral, intervalo de longitud
de onda que contenga las longitudes de los analitos a analizar y provisto de
una interfase con registrador o un adecuado sistema de datos.
5.1.3 Generador de
Hidruros
5.1.4 Accesorio de vapor
frío para la determinación de mercurio
5.1.5 Horno de grafito:
Equipo capaz de programar las temperaturas, tiempos y flujos de gas inerte y
alterno en los pasos para atomizar la muestra, debe contar con un sistema de
enfriamiento para controlar la temperatura del horno.
5.1.6 Lámparas de:
arsénico, cadmio, cromo, cobre, cobalto, estaño, manganeso, mercurio, níquel,
plomo, selenio y zinc.
5.1.7 Horno de microondas
y/o placa de calentamiento para digestión de muestras ambientales.
5.1.8 Quemadores de 10 cm
de 1 ranura y para óxido nitroso de 5 cm o los recomendados por el fabricante
del equipo.
5.1.9 Celda de cuarzo en
forma de “T” para el generador de hidruros o la recomendada por el fabricante
del equipo.
5.2 Materiales
Todo
el material volumétrico utilizado en este método debe ser clase A, demostrar su
calibración con un certificado o bien a través de un procedimiento de
verificación de la calibración.
5.2.1 Limpieza del
Material
a) Todo el
material usado en esta determinación debe ser exclusivo para este método.
Remojar durante una hora en una disolución de ácido nítrico al 10% el material
y enjuagar con agua. No se permite el uso de detergentes con base de amoniaco
para la limpieza del material.
b) Los contenedores de las
muestras deben lavarse con disolución de detergente libre de metales,
enjuagarse con agua, remojarse en disolución de ácido toda la noche y volver a
enjuagarse con agua libre de metales, dejar secar (con cuidado especial para el
análisis de trazas).
c) En los casos
de que el material presente grasas, enjuagar con acetona y/o hexano.
5.2.2 Papel filtro
número 40 (o equivalente).
5.2.3 Pipetas
volumétricas tipo A o micropipetas calibradas.
5.2.4 Cajas de Petri.
5.2.5 Tubos de grafito y
accesorios (específicos para el horno de grafito utilizado).
5.2.6 Material de consumo
que necesite el espectrofotómetro en flama y/o horno y/o generador de hidruros
y/o vapor frío.
5.2.7 Membranas de
filtración de 0,45 micras.
6. Reactivos y patrones
Todos
los reactivos utilizados en este método deben ser grado reactivo, para las
determinaciones que se realicen en horno de grafito deben ser grado suprapuro o
equivalente.
Agua:
Debe cumplir con las siguientes características:
|
|
Agua Tipo I |
Agua Tipo II |
|
Resistividad
(megaohm-cm a 25ºC) |
18 |
>10 |
|
Conductividad
(S/cm a 25ºC) |
=
0.06 |
<
0.1 |
6.1 Acido clorhídrico concentrado
(HCl).
6.2 Acido nítrico concentrado
(HNO3).
6.3 Acido sulfúrico
concentrado (H2SO4).
6.4 Aire comprimido libre de
agua y aceite.
6.5 Acetileno grado absorción
atómica.
6.6 Argón grado alta pureza o
absorción atómica.
6.7 Nitrógeno grado alta
pureza o absorción atómica.
6.8 Oxido nitroso grado alta
pureza o absorción atómica.
6.9 Disolución de borohidruro
de sodio (NaBH4) en hidróxido de sodio (NaOH) a la concentración
especificada por el fabricante del equipo. Esta disolución debe prepararse
justo antes de realizar el análisis.
6.10 Disolución patrón
(certificada y/o preparada en el laboratorio de una concentración de 1 g/l 1
g/Kg) de los siguientes metales: arsénico, cadmio, cromo, cobre, estaño,
manganeso, mercurio, níquel, plomo, selenio y zinc.
6.11 Disolución patrón
intermedia: Preparar las disoluciones patrón de acuerdo al método tomando una
alícuota adecuada de la disolución patrón certificada.
6.12 Disoluciones patrón:
Demostrar el intervalo lineal con un mínimo de 4 disoluciones y un blanco que
estén dentro del intervalo de trabajo.
6.13 Disolución patrón para la
matriz adicionada: La concentración depende del metal a analizar y de la
técnica utilizada, se debe preparar a partir de la disolución patrón
intermedia.
6.14 Disolución de cloruro de
sodio con sulfato de hidroxilamina: Disolver 12,0 g de NaCl y 12,0 g de
hidroxilamina en agua, aforar a 100 ml.
6.15 Disolución de cloruro
estañoso al 10% en ácido clorhídrico al 20% o como lo indique el fabricante.
7. Recolección,
preservación y almacenamiento de muestras
7.1 Las muestras
deben recolectarse y almacenarse en frascos de polietileno de alta densidad,
correctamente identificados y cerrados para evitar pérdidas de materiales
volátiles durante la transportación hacia el laboratorio.
7.2 Las muestras
deben conservarse a una temperatura de 4ºC.
7.3 No se debe
adicionar ningún preservador a las muestras
7.4 El tiempo
máximo para el análisis es de 28 días
8. Control de calidad
8.1 Aspectos
generales
8.1.1 Cada
laboratorio que utilice este método está obligado a operar un programa de
control de calidad (CC) formal.
8.1.2 El desempeño del laboratorio se debe comparar con los criterios
establecidos en la Sección de Desempeño, con objeto de determinar si los
resultados de los análisis cumplen con las especificaciones del método.
8.1.3 El analista
debe hacer una demostración inicial de su habilidad para generar una exactitud
y precisión aceptables por este método. El procedimiento debe realizarse como
se menciona en la Sección de demostración inicial de la capacidad del
laboratorio.
8.1.4 Cada vez que
se realice una modificación al método o que se cambie al analista responsable
de llevar a cabo esta determinación, el analista designado debe repetir el
procedimiento mencionado en la Sección de desempeño inicial del método, si el
cambio va a afectar alguno de los parámetros de desempeño del método, el
laboratorio debe demostrar que los nuevos parámetros determinados son iguales o
mejores que los anteriores.
8.1.5 No se permite
el uso de técnicas determinativas alternativas y cambios que degraden la
ejecución del método. Si se utiliza una técnica analítica que no sea la
especificada en este método, dicha técnica debe tener especificaciones iguales
o mejores que la de la técnica descrita en este documento para el analito de
interés.
8.1.6 Es
obligatorio para el laboratorio mantener los registros de las modificaciones
realizadas a este método. Estos registros deben de incluir lo siguiente:
- La justificación por
escrito de la necesidad de realizar modificaciones al método para ese analito.
- Resultados de todas las
pruebas de CC comparadas del método modificado con el método original, dichos
datos deben de incluir todos los parámetros mencionados en la Sección de
Desempeño del Método.
-Información que permita a
un evaluador externo validar cada determinación mediante el seguimiento de la
información desde la recepción de la muestra hasta el resultado final. Lo
anterior debe estar debidamente registrado e incluir, al menos los siguientes
puntos:
a)
Identificación de la muestra
b) Número del
lote analítico en el cual se analizó la muestra
c) Fecha del
análisis
d) Procedimiento
cronológico utilizado
e) Cantidad de
muestra utilizada
f) Número de
muestras de control de calidad analizadas en el lote
g) Trazabilidad
de las calibraciones de los instrumentos de medición
h) Registros de
bitácoras, en cintas magnéticas o en otros respaldos de información
i) Información
cruda reportada por los equipos o por los analistas
j) Evidencia de la
aceptación o rechazo de los resultados del lote analítico
k) Los nombres,
títulos, direcciones y número de teléfono de los analistas que ejecutaron los
análisis y modificaciones y el encargado del control de calidad que presenció y
verificó los análisis y sus modificaciones.
8.2 Demostración
inicial de la capacidad del laboratorio:
8.2.1 Verificación
de la exactitud de la calibración inicial del método.- Se debe verificar la
curva de calibración con patrones de procedencia diferente a los utilizados para
la curva de calibración y elaborados por personal diferente al analista
encargado, con objeto de validar que la curva de calibración está adecuadamente
elaborada.
8.2.1 Los valores
entre los obtenidos en la curva de calibración y los patrones de procedencia
diferente deben variar menos de 10%.
8.2.2 Si los
valores no cumplen con lo especificado, determinar las causas y corregir los
errores, documentar adecuadamente las incidencias y acciones correctivas e
incluirlas en el expediente de desempeño inicial del método (EDIM).
8.2.3 Repetir el
procedimiento anterior hasta que se cumpla con la especificación.
8.3 Límite de
detección del método (LDM):
8.3.1 Preparar una
muestra sintética a partir del patrón de calibración más adecuado a una
concentración que se encuentre entre 5 y 10 veces el Límite de Detección del
Método estimado.
8.3.2 Dividir la
disolución anterior en 7 alícuotas y analizarlas.
8.3.3 Calcular el
promedio y la desviación estándar de los resultados obtenidos.
8.3.4 Calcular el
LDM usando la siguiente ecuación:
LDM = (tn-1, 0.99%)*s
Donde:
(tn-1, 0.99%)
= 3.14
s = desviación estándar
8.3.5 Si el
resultado del LDM obtenido es menor de 10 veces la concentración de la
disolución patrón que se utilizó, entonces preparar otra disolución que se
encuentre entre 5 y 10 veces el LDM obtenido y repetir el procedimiento a
partir del inciso 8.3.
8.3.6 El
procedimiento y los resultados deben quedar asentados en la bitácora del
analista y en el Expediente del Desempeño Inicial del Método (EDIM).
8.3.7 El Límite de
Detección del Método que se obtenga, debe ser igual o menor al que se presente
en la Sección de Desempeño.
8.4 Intervalo de
Trabajo del Método (ITM): El ITM se debe centrar de acuerdo al Límite Máximo
Permisible de la Norma Oficial Mexicana respectiva, con al menos dos puntos de
la curva de calibración por debajo de dicho valor y dos puntos por encima.
8.4.1 El intervalo
de trabajo se define como la porción lineal de la curva de calibración,
normalmente abarca de 1 a 3 órdenes de magnitud.
8.4.2 Se puede
determinar utilizando como indicador el coeficiente de correlación de la recta
de ajuste por mínimos cuadrados, el cual debe ser mayor a 0,99 (siempre y
cuando se verifique la linealidad por métodos gráficos).
8.5 Exactitud
Inicial del Método.- Calcular la exactitud inicial del método de la siguiente
forma:
8.5.1 Preparar a
partir de un material de referencia, la concentración intermedia del analito de
interés dentro de su intervalo de trabajo, divida en 10 porciones la muestra y
realice el análisis completo en las mismas condiciones de operación y por el
mismo analista.
8.5.2 Analizar las
10 muestras y registrar los resultados.
8.5.3 Calcular el
por ciento de recuperación:
%R
= (Valor Encontrado/Valor Real)* 100
Donde:
%R = Por ciento de Recuperación
Valor Encontrado Valor
medido de la muestra
Valor Real= Valor asignado
a la muestra
8.5.4 Calcular el
promedio y la desviación estándar del %R.
8.5.5 Comparar los
valores de la media y la desviación estándar del %R con los que se presentan en
la Sección de Desempeño del Método.
8.5.6 Si los
valores no cumplen con lo especificado, determinar las causas y corrija los
errores, documentar adecuadamente las incidencias y acciones correctivas e
inclúyalas en el EDIM.
8.5.7 Repetir el
procedimiento anterior hasta que se cumpla con las especificaciones de los
criterios de aceptación de la Sección de Desempeño del Método.
8.6. Precisión
inicial del método.- Calcular la precisión inicial del método de la siguiente
forma:
8.6.1 Con los
resultados de las 10 muestras preparadas en la Sección anterior elabore una
tabla donde se pongan los 10 valores apareados (5 renglones con 2 valores
apareados).
8.6.2 Calcular la
diferencia porcentual relativa (DPR) con la siguiente ecuación:
DPR
= 200 (X1 - X2)/(X1 + X2)
Donde:
DPR= Diferencia Porcentual
Relativa
X1 = Valor
medido de la muestra original
X2 = Valor
medido de la muestra duplicada.
8.6.3 Calcular la
media aritmética del DPR.
8.6.4 Comparar los
valores de la media con los que se presentan en la Sección de Desempeño del
Método.
8.6.5 Si los
valores no cumplen con los especificados, determinar las causas y corrija los
errores, documentar adecuadamente las incidencias y acciones correctivas e
inclúyalas en el EDIM.
8.6.6 Repetir el
procedimiento anterior hasta que se cumpla con las especificaciones de los
criterios de aceptación de la Sección de Desempeño del Método.
8.7 Límite
Práctico de Cuantificación (LPC)-Se puede calcular multiplicando por 5 el LDM o
en caso de que el rango de trabajo sea mayor a este valor, entonces utilizar el
primer punto de la curva de calibración.
8.8 Cada lote
analítico deberá estar compuesto de la siguiente forma:
1 Muestra de Verificación del
Instrumento (MVI).
2 Muestra de Verificación de
la Calibración Inicial (MVCI) (o patrones de la curva de calibración).
3 Blanco de Reactivos (BR)
4 Muestra Sintética de
Control de Calidad (MCC)
5 Muestra real No. 1
6 Muestra real No. 2
7 Muestra real No. 1
duplicada (MD)
8
a 13 Muestras reales Nos. 3 a 10
14 Muestra de Verificación de
la Calibración Continua (MVCC)
15 MCC No. 2
16 Muestra real No. 11
17 Muestra real No. 12
18 Muestra real No. 11 duplicada (MD)
19
a 26 Muestras reales Nos. 13 a
20, etc.
Para lotes mayores, debe
analizarse al menos un 10% de MCC y 10% de MDs.
8.9 Muestras de
Control de Calidad
8.9.1 Blanco de
Reactivos (BR): Es una matriz libre de analitos a la cual se le agregan todos
los reactivos en los mismos volúmenes o proporciones usados en el procesamiento
de la muestra. El BR debe llevarse a través de la preparación de la muestra y
el procedimiento analítico. El BR se usa para documentar la contaminación
resultante del proceso analítico.
8.9.1.1 Para que un
BR se considere adecuado, la concentración en el mismo de cualquier analito no
deberá ser más alto que el LPC correspondiente.
8.9.1.2 Nunca se
deberá sustraer el valor del BR al de las muestras o calibraciones analizadas.
8.9.1.3 Si el valor
del BR es mayor al LPC se deberá desechar el lote analítico, determine las
causas y corrija los errores, documente adecuadamente las incidencias y
acciones correctivas en la bitácora del analista.
8.9.2
Muestras Duplicadas (MD): Son muestras reales o de control de calidad que se
preparan a partir de una misma muestra, la variación entre ellas sólo es debida
al error aleatorio de la pareja analista-método.
8.9.3
Muestras Sintéticas de Control de Calidad (MCC): Son muestras preparadas a
partir de agua reactivo y patrones de referencia que se utilizan para
determinar la exactitud y precisión de la pareja analista-método.
8.9.3.1 Las
MCC se deben preparar a una concentración aproximada al valor medio de la curva
de calibración.
8.9.3.2
Deben ser preparadas a partir de patrones diferentes a los que se utilizaron
para la curva de calibración y por personal diferente.
Nota: Se
recomienda que el analista no conozca el valor de las soluciones de MCC con el
objeto de asegurar la veracidad de la medición.
8.9.4
Muestra de Verificación de la Calibración Inicial (MVCI): Se utiliza para
verificar que la curva de calibración sigue vigente a través de diferentes días
de trabajo.
8.9.4.1 Se
debe utilizar la disolución patrón de valor intermedio de la curva de
calibración y analizarla después de haber verificado el instrumento de
medición.
8.9.4.2 La
variación máxima permitida es de 25%, si el valor
encontrado, es mayor, entonces debe realizar una nueva curva de calibración.
8.9.5
Muestra de Verificación de la Calibración Continua (MVCC): Se utiliza para
verificar que la curva de calibración sigue vigente a través del mismo día de
trabajo.
8.9.5.1 Se
debe utilizar la disolución patrón de valor intermedio de la curva de
calibración y analizarla cada 10 muestras dentro del lote analítico.
8.9.5.2 La
variación máxima permitida es de 20%, si el valor
encontrado, es mayor, entonces debe realizar una nueva curva de calibración,
analizar de nuevo las 5 muestras anteriores y proseguir con el análisis del
lote analítico.
8.9.6
Muestra de Verificación del Instrumento (MVI): Es la muestra que sirve para
verificar que el instrumento de medición se encuentra en las condiciones
apropiadas de funcionamiento y que son las mismas en las cuales se realizó la
calibración inicial.
8.9.6.1 La
MVI es una muestra que depende de las especificaciones del fabricante del
instrumento.
8.10
Control de Calidad Estadístico.- En esta sección se especifica cómo debe
realizarse el control de calidad estadístico obligatorio para este método:
8.10.1
Gráficas de Control de Exactitud.- El laboratorio debe elaborar y mantener
actualizadas las gráficas de control de exactitud para cada lote analizado a
partir de la demostración inicial de desempeño. Para poder iniciar la gráfica,
es necesario contar con al menos 12 datos de muestras de MCC, antes de tener
este número de datos, se pueden utilizar como criterio de aceptación y rechazo
los límites encontrados en el estudio de desempeño inicial del método. Para
elaborar la gráfica de control de exactitud deberá utilizarse el siguiente
procedimiento:
8.10.2
Calcular el por ciento de recuperación de acuerdo a la siguiente ecuación:
%R = (Valor Encontrado/Valor Real)* 100
Donde:
%R = Por ciento de Recuperación
Valor Encontrado = Valor
medido de la muestra de CC
Valor Real = Valor asignado a la muestra de CC.
8.10.3 Con
al menos 12 datos, calcular la media aritmética (X) y la desviación estándar para el %R.
8.10.4 Los
límites de control son los siguientes:
a)
Límite de control superior = X + 2s
b)
Límite de advertencia superior = X + 1s
c)
Límite de control inferior = X-2s
d)
Límite de advertencia inferior = X-1s
8.10.5 Representar una gráfica de control, dibujando una
línea paralela al eje de las abscisas (X), representando la media como una
línea central y líneas paralelas a la línea central, representando los límites
de control y los límites de advertencia superior e inferior.
8.10.6 Cada
valor de exactitud obtenido de las MCC de cada lote analizado deberá graficarse
y estar dentro de los límites de control superior e inferior.
8.10.7 Si
un valor de exactitud es mayor a 2s, deberán
rechazarse todos los resultados del lote analítico, determine las causas y
corrija los errores, documente adecuadamente las incidencias y acciones
correctivas en la bitácora del analista.
8.11
Gráficas de Control de Precisión.- El laboratorio debe elaborar y mantener
actualizadas las gráficas de control de precisión para cada lote analizado a
partir de la demostración inicial de desempeño. Para poder iniciar la gráfica,
es necesario contar con al menos 24 datos de muestras duplicadas, antes de
tener este número de datos, pueden utilizarse como criterio de aceptación y
rechazo los límites encontrados en el estudio de desempeño inicial del método.
Para elaborar la gráfica de control de precisión deberá utilizarse el siguiente
procedimiento:
8.11.1
Calcular la Diferencia Porcentual Relativa de acuerdo a la siguiente ecuación:
DPR = 200 /X1 - X2/ /(X1 + X2)
Donde:
DPR = Diferencia Porcentual
Relativa
X1 = Valor medido de la muestra original
X2 = Valor medido de la muestra duplicada.
/X1 - X2/ = Valor
absoluto de la diferencia de los dos datos
8.11.2 Con
al menos 12 datos de la DPR, calcular el promedio.
8.11.3
Determinar los límites de control de la siguiente forma:
Límite Superior de Control (LSC) = 3.27R
Límite Superior de Advertencia (LSA) = 2.51R
Donde:
R = Promedio de las DPR calculadas
8.11.4
Representar en una gráfica de control líneas paralelas al eje-X, los límites
superiores de advertencia y límites superiores de control (LSA y LSC).
8.11.5 Cada
valor del DPR obtenido de las muestras duplicadas de cada lote analizado deberá
graficarse y ser menor que el LSC.
8.11.6 Si
un valor de precisión es mayor a las especificaciones mencionadas en el inciso
anterior, deberán rechazarse todos los resultados del lote analítico,
determinar las causas del problema y corregir los errores, documentar
adecuadamente las incidencias y acciones correctivas e incluirlas en la
bitácora del analista.
8.12
Validación de modificaciones del método o de métodos alternos.- Para validar
las modificaciones que se efectúen a este método o para la utilización de
métodos alternos deberá seguirse el siguiente procedimiento:
8.12.1 Si
se realizan modificaciones al presente método, deberán validarse de acuerdo a
lo que se presenta en la sección de desempeño inicial del método.
8.12.2 Si
se utiliza un método alterno cuya fuente sea un método estandarizado por alguna
institución de carácter internacional o reconocida internacionalmente (p.e.
ASTM, USEPA, AOAC, Standard Methods, DIN, OMS Environment Canada, etc.) siga el
mismo procedimiento que se presenta en la Sección desempeño inicial del método.
8.12.3 Si
se utiliza algún método no estandarizado, deberá evidenciarse, además de los
parámetros mencionados en la sección de desempeño del método, los parámetros de
Robustez, Reproducibilidad y Especificidad los cuales sólo pueden evaluarse
mediante estudios interlaboratorios.
8.13
Dependiendo de los requerimientos del programa específico de control de calidad
de algún proyecto, pueden requerirse muestras dobles de campo, para evaluar la
precisión y exactitud del muestreo y las técnicas de transportación de la
muestra y otras muestras especiales de control de calidad como muestras
adicionadas y muestras adicionadas duplicadas para verificar las interferencias
de matriz.
9.
Calibración
9.1
Calibración inicial.
9.1.1
Calibrar el equipo de acuerdo a las especificaciones del fabricante.
9.1.2
Preparar la curva de calibración mínimo con cinco niveles de concentración.
9.1.3 Incluir
un blanco de reactivos en cada uno de los lotes de análisis.
9.2
Calibración continua.
9.2.1 Para
verificar la calibración del instrumento en un proceso continuo, analizar un
blanco de calibración y la muestra de verificación del instrumento diariamente
o cada 10 análisis, la muestra de verificación corresponde a un nivel de
concentración de la curva de calibración, si el valor de la concentración
obtenida varía en más del 10% del valor real, el instrumento deberá ser
calibrado de nuevo, así como la respuesta de la muestra de verificación, una
vez que la muestra de verificación cumple con lo especificado, analizar las
muestras previas en grupos de 5 con una solución estándar de concentración
igual al nivel medio de la curva de calibración. Si el valor de la solución
estándar tiene una desviación mayor al 10% con respecto al valor real el
analista debe identificar la fuente del problema y tomar las acciones
correctivas necesarias para solucionar el problema.
10.
Procedimiento
10.1
Preparación de la muestra
10.1.1
Anotar el nivel del líquido en cada uno de los contenedores de las muestras,
determinar si cualquier muestra presentó pérdida durante el transporte, si se
trata de una cantidad considerable de fuga, se debe informar al cliente de esta
pérdida y él debe autorizar la corrección para el cálculo del resultado final.
La figura número 2 ilustra los procedimientos para la preparación y el análisis
de la muestra para cada uno de los componentes del tren de muestreo.
10.1.2
Contenedor número 1 (Filtro de la muestra)
10.1.2.1 Si
se determinan emisiones de partículas, primero desecar el filtro y el colector
del filtro a temperatura ambiente (no calentar los filtros para acelerar el
secado) hasta obtener un peso constante.
10.1.2.2 Si
no se determinan emisiones de partículas, dividir el filtro y su colector en
porciones de 0,5 g aproximadamente cada una, digerir las muestras según el
procedimiento de digestión.
10.2.2
Contenedor número 2 (Solución de enjuague de acetona)
10.2.2.1
Registrar el nivel del líquido en el contenedor y confirmar en la hoja de
análisis si se identificó cualquier pérdida durante el transporte, si se trata
de una cantidad considerable de fuga se debe informar al cliente de esta
pérdida y él debe autorizar la corrección para el cálculo del resultado final.
Medir el líquido en este contenedor
volumétricamente con una precisión de 1,0 ml o
gravimétricamente con una precisión de 0,5 g, transferir
el contenido a un vaso de 250 ml libre de ácido, evaporar hasta sequedad a
temperatura y presión ambiente, si se determinan emisiones de partículas
desecar por 24 horas sin exponer a calor, pesar hasta obtener peso constante,
reportar el resultado con una precisión de 0,1 mg. Disolver el residuo
remanente con 10 ml de HNO3 concentrado,
cuantitativamente combinar la muestra resultante (incluyendo todos los líquidos
y cualquier partícula), con el contenedor número 3 antes de iniciar el
procedimiento de la sección siguiente.
10.2.3 Contenedor
número 3 (Solución de enjuague de las sonda)
10.2.3.1
Verificar que el pH de esta muestra sea igual o menor de 2,0 unidades de pH, si
no es así acidificar la muestra adicionando HNO3 concentrado hasta obtener un pH de 2,0.
Enjuagar la muestra con agua dentro de un vaso, digerir la muestra según el
procedimiento de digestión, mezclar la muestra digerida resultante con las
porciones del ácido digerido de la preparación del filtro previamente descrita
en la sección 10.1.2.1, después mezclar la muestra resultante directamente con
las porciones del ácido digerido de la preparación en la sección 10.2.2.1. La
muestra resultante se identificará como la “fracción de la muestra 1”.
10.2.3.2
Filtrar la muestra combinada con un filtro de papel número 541. Diluir con agua
a 300 ml aproximadamente, esta muestra se identificará como “fracción analítica
1”, medir y registrar el volumen de la fracción analítica 1 con una precisión
de 0,1 ml, cuantitativamente transferir una alícuota de 50 ml e identificarla
como “fracción analítica 1B”, identificar la porción restante como “fracción
analítica 1A”, esta fracción es utilizada para analizar todos los metales
excepto mercurio. La fracción analítica 1B es la que se utiliza para la
determinación de mercurio.
10.2.4 Contenedor
número 4 (Impactores 1 -3)
10.2.4.1
Medir y registrar el volumen total de esta muestra con una precisión de 0,5 ml
e identificarla como “fracción de la muestra número 2”, tomar de ésta una
alícuota de 75 a 100 ml e identificarla como “fracción de la muestra 2 B”,
identificar la porción sobrante como “fracción de la muestra 2A”. La fracción
de la muestra 2A define el volumen de la fracción analítica 2A antes de la
digestión. Someter a digestión la fracción de la muestra 2A, una vez digerida
identificar ésta como “Fracción analítica 2A”, el volumen de ésta es
aproximadamente de 150 ml y se utiliza para la determinación de los metales
incluidos en esta norma a excepción del mercurio.
10.2.4.2
Verificar el pH de la fracción de la muestra 2A que debe ser menor o igual a
2,0 unidades de pH, en caso de ser necesario ajustar el pH con HNO3 concentrado.
10.2.4.3
Digerir la fracción de la muestra 2A según procedimiento de digestión.
10.2.5
Contenedor 5A (impactor número 4), Contenedores números 5B y 5C (impactores 5 y
6)
10.2.5.1
Mantener separadas las muestras de los contenedores 5A, 5B y 5C.
10.2.5.2
Medir y registrar el volumen del contenedor 5A, con una precisión de 0,5 ml,
identificar éste como “fracción analítica 3A”.
10.2.5.3
Filtrar el contenido del contenedor 5B a través de un filtro de papel del
número 40, esto con el fin de remover cualquier precipitado que se halla
formado a partir del MnO2,
recuperar el filtrado en un matraz volumétrico de 500 ml, aforar hasta la marca
con agua destilada. Identificar este filtrado como “fracción analítica 3B”.
Conservar el papel filtro que se usó en la filtración para su posterior
análisis.
10.2.5.4
Digestión del filtro usado en la sección 10.2.5.3, transferir el filtro a un
recipiente apropiadamente ventilado, colocar este recipiente en la campana de
extracción, adicionar 25 ml de una solución de HCl 8,0 N, iniciar la digestión
por un tiempo mínimo de 24 horas a temperatura ambiente.
10.2.5.5
Filtrar el contenido del contenedor 5C con un filtro de papel del número 40,
recuperar el filtrado en un matraz volumétrico de 500 ml, en este mismo matraz
filtrar el producto de digestión de la sección 10.2.5.4.
10.2.6
Contenedor número 6 (Sílica gel)
10.2.6.1
Pesar en balanza analítica la sílica gel gastada (o sílica gel más el impactor)
con una precisión de 0,5 g. Registrar
este dato.
10.3
Procedimiento de digestión convencional de disoluciones y filtros de cuarzo
10.3.1
Adicionar 30 ml de una solución de HNO3 al 50% v/v al vaso de precipitado que
contiene la muestra a digerir, calentar durante 30 minutos aproximadamente
sobre una placa de calentamiento, controlando la temperatura para mantenerla
por debajo de la temperatura de ebullición.
10.3.2
Adicionar 10 ml de una solución de peróxido de hidrógeno al 3% v/v, calentar
por 10 minutos más.
10.3.3
Adicionar 50 ml de agua caliente y dejar calentar la muestra por 20 minutos
más.
10.3.4
Dejar enfriar la muestra y aforar a 150 ml con agua destilada (o a un volumen
apropiado según la concentración de metales).
10.3.5
Digestión en horno de microondas para filtros de cuarzo:
10.3.5.1
Reactivos
- Acido nítrico HNO3 al 69%, grado suprapuro
- Acido bórico H3BO3, grado reactivo
- Acido fluorhídrico al 49%, grado reactivo
10.3.5.2
Disoluciones
- Disolución saturada de ácido bórico.- En un
vaso de precipitado con aproximadamente 500 ml de agua, disolver la cantidad
suficiente de ácido bórico hasta la sobresaturación (mantener la disolución en
agitación moderada hasta asegurar la saturación).
10.3.5.3
Equipos
- Horno de microondas capaz de censar la
presión en PSI y de alcanzar hasta 200 PSI.
10.3.5.4
Procedimiento
a)
Colocar el filtro de cuarzo que contenga la muestra en un vaso de PTFE,
adicionar 5,0 ml de agua ASTM I, posterior adicionar 1,0 ml de ácido
fluorhídrico y 5,0 ml de ácido nítrico, utilizar el siguiente programa de
digestión:
1. Cinco minutos a 60 PSI
2. Cinco minutos a 120 PSI
3. Cinco minutos a 160 PSI
4. Cinco minutos a 190 PSI
5. A una potencia de 630 watts para 6 vasos.
b) Dejar enfriar el sistema de 30 a 45 minutos.
c) Adicionar 5,0 ml de disolución de ácido bórico, para
neutralizar el excedente de ácido fluorhídrico.
d) Digerir de nuevo las muestras a 100 PSI,
durante cinco minutos a una potencia de 630 watts para seis vasos.
e) Completada la digestión dejar enfriar el
sistema y los vasos.
f) Aforar con agua destilada tipo ASTM I, en matraces de
polipropileno a un volumen de 25 o 50 ml.
10.3.5.5 Como
parte del programa de control de calidad se debe incluir un filtro blanco en el
proceso de digestión.
10.4
Procedimiento para digestión de muestras para análisis de mercurio.- Las
muestras que se sometan a este análisis se deben digerir por el método
convencional de digestión mencionado en la sección 10.3 y posteriormente someterse al siguiente procedimiento:
10.4.1
Adicionar a la muestra 2,0 ml de H2SO4 concentrado.
10.4.2
Adicionar 1,0 ml de HNO3
concentrado.
10.4.3
Adicionar una solución de permanganato de potasio al 5%, hasta que permanezca
el color, dejar reposar por 15 minutos para asegurar la oxidación total.
10.4.4
Adicionar la solución de persulfato de potasio al 5%, calentar en baño maría a
95ºC por dos horas aproximadamente. Enfriar a temperatura ambiente.
10.4.5
Adicionar la cantidad necesaria de la solución de hidroxilamina para reducir el
exceso de permanganato (hasta decoloración de la muestra).
10.4.6
Adicionar la solución de cloruro estañoso para liberar el mercurio en estado
basal. El vapor atómico de mercurio es llevado a la celda de absorción para su
detección.
10.5
Identificación de las muestras
10.5.1 Para
cada tren de muestreo corrido, se deben obtener siete muestras, de las cuales
dos se usan para el análisis de todos los metales y cinco para el análisis de
mercurio. La identificación esquemática de cada contenedor de la muestra, la
preparación analítica descrita y el esquema del análisis se presentan en la
Figura 2.
10.5.2 La
primera muestra identificada como fracción analítica 1A y 1B, corresponden a la
muestra digerida en la mitad anterior del tren. En la fracción analítica 1B
sólo se analizará mercurio.
10.5.3 La
tercera y cuarta muestras analíticas identificadas como fracción analítica 2A y
2B contienen las muestras de la remoción de humedad del impactor número 1 (si
se usa) y de los impactores números 2 y 3 que contienen HNO3/H2O2.
10.5.4 La
fracción analítica 2A es utilizada para el análisis de blancos de los metales a
excepción de mercurio, la fracción analítica 2B se utiliza para el análisis de
mercurio.
10.5.5 La
quinta, sexta y séptima muestras (identificadas como fracciones analíticas 3A,
3B y 3C) corresponden a los contenidos de los impactores 5 y 6 de H2SO4/KMnO4, así como al del
impactor número 4 y a las soluciones de enjuague.
10.5.6 La
mitad posterior del mercurio total se determina en la suma de las fracciones
analíticas 2B, 3A, 3B y 3C. Las fracciones 1A y 2A se pueden combinar
proporcionalmente antes de ser analizadas.
10.5.7 La determinación
de mercurio se realizará en las fracciones analíticas 1B, 2B, 3B y 3C.
Nota: Para cada muestra
original, seleccionar una alícuota en el intervalo de 1 a 10 ml, si no se
conoce la cantidad de mercurio esperada en la muestra, se recomienda tomar una
alícuota de 5,0 ml para la primera dilución a 100 ml. La concentración total de
mercurio en la alícuota debe ser menor a 1 g y estar dentro
del intervalo de cero a 100 ng de la curva de calibración.
10.5.8 Colocar la
alícuota de la muestra en una botella de DBO por separado, adicionar suficiente
agua para obtener un volumen total de 100 ml, continuar con la digestión y
preparación de la muestra como se indica en la digestión para mercurio.
10.6 Análisis de las
muestras
10.6.1 Análisis por aspiración directa (aire-acetileno y/o
óxido nitroso-acetileno).
10.6.1.1
Encender el equipo y conectar la lámpara para el metal que se va a determinar.
10.6.1.2 Alinear la lámpara hasta obtener la máxima energía.
10.6.1.3 Seleccionar el
ancho de la banda espectral óptimo, el cual depende de cada elemento en
particular.
10.6.1.4 Seleccionar la
longitud de onda para el metal de interés de acuerdo al manual del fabricante o
bien a la tabla anexa número 16.1.2.
10.6.1.5 Optimizar la
longitud de onda ajustándola hasta obtener la máxima energía.
10.6.1.6 Esperar de 10 a 20
minutos para que se estabilice el equipo, una vez encendida la lámpara.
10.6.1.7 Ajustar las
condiciones de la flama aire-acetileno de acuerdo a las indicaciones del
fabricante. Encender la flama, permitir que el sistema alcance el equilibrio de
temperatura.
10.6.1.8 Aspirar un blanco
de reactivos (matriz libre de analitos a la cual se le agregan todos los
reactivos en los mismos volúmenes y proporciones usadas en el procesamiento de
la muestra).
10.6.1.9 Aspirar una
disolución patrón del metal a analizar y ajustar la velocidad de flujo del
nebulizador hasta obtener la máxima sensibilidad. Así como ajustar el quemador
horizontal y verticalmente hasta obtener la máxima respuesta.
10.6.1.10 Analizar la curva
de calibración con un mínimo de cinco concentraciones y un blanco de reactivos
en el intervalo lineal demostrado para cada elemento. El primer punto deberá
ser igual o mayor al límite de cuantificación, y el último deberá estar dentro
del intervalo lineal.
10.6.1.11 Representar los
valores obtenidos de las absorbancias contra las concentraciones en una gráfica
y realizar los cálculos cuantitativos. Analizar el lote de muestras.
10.6.2 Análisis por
Generador de Hidruros:
10.6.2.1 Instalar la lámpara
adecuada, colocar la corriente de la lámpara dependiendo del metal a analizar.
10.6.2.2 Encender el
espectrofotómetro y esperar a que se estabilice.
10.6.2.3 Seleccionar la
longitud de onda y el ancho de banda espectral para el elemento que va a ser
determinado de acuerdo al protocolo del laboratorio o del manual del
fabricante.
10.6.2.4 Alinear la lámpara
a su máxima energía.
10.6.2.5 Alinear el
accesorio que se va a usar para atomizar la muestra.
10.6.2.6 Ajustar el rayo de
luz de la lámpara de acuerdo con las especificaciones del fabricante del
equipo.
10.6.2.7 Ajustar los flujos
de gas de aire y acetileno. Este ajuste no se requiere para la determinación de
mercurio.
10.6.2.8 Alinear la celda de
cuarzo en el rayo de luz y esperar de 20 a 30 minutos para su estabilización en
la flama antes de iniciar el análisis; en este periodo, preparar las
disoluciones estándar y los reactivos.
10.6.2.9 Colocar en el
recipiente del reductor una disolución de borohidruro de sodio en hidróxido de
sodio y conectar el recipiente al sistema según las especificaciones del
fabricante del equipo.
10.6.2.10 Abrir el suministro
de gas inerte y ajustar la presión de acuerdo a las especificaciones del
fabricante del equipo.
10.6.2.11
Conectar el vaso de reacción al sistema generador y esperar el tiempo
suficiente para que todo el aire se purgue del sistema, entonces registrar el
cero en el espectrofotómetro (autocero). Existen equipos que cuentan con
sistemas de inyección de flujo en este caso aplicar el procedimiento
recomendado por el fabricante.
10.6.2.12 Conectar el vaso de reacción que contiene el blanco
de reactivos.
10.6.2.13 Purgar
el sistema hasta eliminar completamente el aire, permitir la entrada de la
disolución de borohidruro de sodio, hasta obtener la lectura del blanco.
10.6.2.14 Limpiar
el sistema haciendo pasar agua o ácido clorhídrico diluido.
10.6.2.15 Analizar
la curva de calibración con un mínimo de cinco concentraciones y un blanco de
reactivos en el intervalo lineal demostrado para cada elemento. El primer punto
deberá ser igual o mayor al límite de cuantificación, y el último deberá estar
dentro del intervalo lineal.
10.6.2.16
Representar los valores de las absorbancias obtenidas contra las
concentraciones en una gráfica (curva de calibración) y realizar los cálculos
cuantitativos.
10.6.2.17 Analizar
el lote de muestras.
10.6.3 Análisis
por Horno de Grafito:
10.6.3.1
Establecer la corriente de la lámpara para cada metal, esperar a que se
estabilice el equipo aproximadamente de 10 a 20 minutos.
10.6.3.2
Seleccionar el ancho de banda espectral óptimo, el cual depende de cada
elemento en particular.
10.6.3.3 Colocar
la longitud de onda en el espectrofotómetro para cada metal de interés de
acuerdo al protocolo del laboratorio o del manual del fabricante.
10.6.3.4 Alinear
el tubo de grafito de acuerdo a las indicaciones del fabricante del equipo.
10.6.3.5
Programar el flujo de gas inerte, gas alterno y de agua de enfriamiento de
acuerdo a lo especificado por el fabricante.
10.6.3.6
Seleccionar el programa para cada uno de los metales con las sugerencias
recomendadas por el fabricante del equipo.
10.6.3.7 Inyectar
la cantidad de muestra especificada por el fabricante en el tubo de grafito.
10.6.3.8 Analizar
la curva de calibración con un mínimo de cinco concentraciones y un blanco de
reactivos en el intervalo lineal demostrado para cada elemento. El primer punto
deberá ser igual o mayor al límite de cuantificación, y el último deberá estar
dentro del intervalo lineal.
10.6.3.9 Inyectar
la cantidad recomendada por el fabricante en el tubo de grafito del blanco de
reactivos y la disolución de trabajo más concentrada para optimizar el programa
del horno.
10.6.3.10 Obtener
una gráfica con los valores de la curva de calibración y realizar los cálculos
cuantitativos.
10.6.3.11 Analizar
el lote de muestras.
10.7 Análisis
por Vapor Frío.
10.7.1 Calibrar
el espectrofotómetro de Absorción Atómica con el aditamento de vapor frío.
10.7.2
Estándares, blancos y muestras serán tratadas con ácido nítrico y ácido
sulfúrico en presencia de permanganato de potasio para oxidar todo el mercurio
presente a forma de Hg2+, el
exceso de permanganato de potasio es reducido con cloruro de hidroxilamina.
El mercurio metálico se reduce con cloruro
estañoso y el vapor atómico del mercurio es llevado por medio del sistema
aereador a la celda de absorción para ser detectado.
10.7.3 Obtener
una gráfica con los valores de la curva de calibración y realizar los cálculos
cuantitativos.
10.7.4 Analizar
el lote de muestras.
11. Cálculos
11.1
Elaborar las curvas de calibración de cada metal de acuerdo a las
concentraciones esperadas en las muestras.
11.2 Calcular la concentración de la muestra por
medio de la ecuación de la recta que se obtiene de las curvas de calibración
para cada metal empleando la siguiente ecuación
Ecuación 1:
y = mx +
b
Donde:
y = Absorbancia (señal) de la muestra
m = Pendiente de la curva de calibración
b = Ordenada al origen
12. Seguridad
12.1 No
se ha determinado la carcinogenicidad de todos los reactivos con precisión, por
lo que cada sustancia química debe tratarse como potencialmente peligrosa para
la salud. La exposición a estas sustancias debe reducirse al menor nivel
posible.
12.2 Este método puede no mencionar todas las
precauciones de seguridad asociadas con su uso.
El laboratorio es responsable de mantener un
ambiente de trabajo seguro y un archivo de las normas de seguridad respecto a
la exposición y manejo seguro de las substancias químicas especificadas en este
método. Debe tenerse un archivo de referencia de las hojas de información de
seguridad el cual debe estar disponible a todo el personal involucrado en estos
análisis.
12.3 El
borohidruro de sodio es una sustancia tóxica, flamable y corrosiva.
12.4 Se
requiere el uso de una campana de extracción, ropa de protección, lentes de
seguridad y mascarilla cuando se preparan las soluciones donde las reacciones
entre el disolvente y el soluto son exotérmicas, esto es, óxido de lantano en
solución ácida. Se requieren iguales precauciones cuando se diluyen ácidos
fuertes, debe evitarse el contacto con la piel y vías respiratorias.
12.5 Se
requiere de un sistema de ventilación permanente para eliminar una gran
cantidad de gases calientes y algunas veces tóxicos producidos por el quemador
durante la operación del instrumento. Como el acetileno es un gas flamable
deberán tomarse las precauciones adecuadas cuando se use. Para evitar
explosiones nunca pase el acetileno a través de instalaciones o tuberías de
cobre o aleaciones con alto contenido de cobre (latón, bronce). Si el
espectrofotómetro no está equipado con un escudo protector, el operador deberá
usar lentes de seguridad para atenuar la luz ultravioleta emitida por la flama.
El óxido nitroso es un gas que se usa como anestésico, por lo que el lugar debe
estar bien ventilado.
12.6 Los
gases oxidantes deben separarse de los gases reductores mediante una pared a
prueba de fuego.
12.7 Seguir
cuidadosamente las guías de operación del fabricante del equipo para optimizar
la velocidad del flujo de gas. Si no se emplean las precauciones adecuadas,
puede resultar una combustión peligrosa dentro de la cámara de mezcla de los
gases.
12.8 Para
evitar explosiones en la línea, no permitir que la presión de llegada del
acetileno al instrumento exceda 1,06 Kg./cm2 (15 psi).
12.9 Cuando
se usa óxido nitroso como oxidante, deberá utilizarse una cabeza de quemador de
50,8 mm de diámetro, ya que utilizando una cabeza de 101,6 mm (4-pulgadas)
ocurrirá un regreso de la flama. La flama de óxido nitroso debe encenderse
usando primero una combinación de aire-acetileno y luego cambiar a óxido
nitroso-acetileno. El óxido nitroso nunca debe pasarse a través de líneas que
contengan residuos de aceites o grasas, ya que puede causar una explosión.
12.10 Revisar
que el tubo del desagüe de la cámara de mezcla de gas esté lleno con agua antes
de comenzar cualquier análisis. Se recomienda el uso de una trampa de seguridad
o de cualquier válvula. Siga las instrucciones del fabricante para mantener una
presión positiva en el sello del líquido.
12.11 Dada la
alta toxicidad del plomo, cadmio, níquel, mercurio y cromo, así como todos los
pasos de preparación y digestión de las muestras, extremar las precauciones de
manejo de todas las disoluciones y utilizar un cuarto bien ventilado.
12.12 El
arsénico, el selenio y sus correspondientes hidruros son tóxicos. Manéjese con
cuidado.
12.13 Los
compuestos del antimonio son irritantes para la piel y las membranas de las
mucosas.
12.14 La
inhalación de los vapores de manganeso han sido reportados como tóxicos para el
ser humano.
13. Desempeño del método
13.1
Límite de detección.- Ver tabla 16.1.1.
13.2
Intervalo de trabajo.- Se debe establecer de acuerdo a los límites máximos
permisibles de cada contaminante establecido en la Norma Oficial Mexicana
correspondiente.
13.3
Precisión inicial del método.- Ver tabla 16.1.1.
13.4
Exactitud inicial del método.- No establecido.
14.
Manejo de residuos
Es la responsabilidad del laboratorio cumplir
con todos los reglamentos federales, estatales y locales referente al manejo de
residuos, particularmente las reglas de identificación, almacenamiento y
disposición de residuos peligrosos.
14.1 Cada
laboratorio debe contemplar dentro de su Programa de Control de Calidad el
destino final de los residuos generados durante la determinación.
14.2
Todas las muestras que cumplan con la Norma de descarga a alcantarillado pueden
descargarse en el mismo sistema.
15. Bibliografía
15.1
“Determination of metals emissions from Stationary Sources”, Emission
Measurement Centers Technical Information EMTIC TM-029, Environmental
Protection Agency, March 22, 1999.
16. Tablas y figuras
Tabla
16.1.1 Límites de detección para análisis por aspiración
directa en espectrofotometría de absorción atómica:
|
Analito (metal) |
Límite de detección en g/l |
|
Arsénico |
2,0 |
|
Cadmio |
5,0 |
|
Cobalto |
50,0 |
|
Cobre |
20,0 |
|
Cromo |
50,0 |
|
Estaño |
|
|
Manganeso |
10,0 |
|
Níquel |
40,0 |
|
Plomo |
100,0 |
|
Selenio |
2,0 |
|
Zinc |
5,0 |
|
Mercurio |
0,02
a 0,2 (depende del volumen que se tome para la digestión y para el análisis) |
Referencia.-
Serie SW846, método 700 EPA.
Tabla
16.1.2 Longitud de onda recomendado para cada analito
|
Analito (metal) |
Longitud de onda |
|
Arsénico |
193,7 |
|
Cadmio |
228,8 |
|
Cobalto |
240,7 |
|
Cobre |
324,7 |
|
Cromo |
357,9 |
|
Estaño |
235,5 |
|
Manganeso |
279,5 |
|
Níquel |
352,4 |
|
Plomo |
283,3 |
|
Selenio |
196,0 |
|
Zinc |
213,9 |
|
Mercurio |
253,7 |
Referencia.-
Serie SW846, método 700 EPA.
16.2 Esquema
de la preparación y análisis de las muestras
ANEXO 4
PROCEDIMIENTO
DE MUESTREO EN FUENTES FIJAS PARA LA DETERMINACIÓN DE METALES
ÍNDICE
1. Objetivo y campo de aplicación
2. Referencias
3. Resumen
4. Interferencias
5. Definiciones
6. Equipo
y materiales
7. Reactivos y patrones
8. Preservación y almacenamiento
de muestras
9. Control de calidad
10. Procedimiento
11. Cálculos
12. Seguridad
13. Manejo
de residuos
14. Bibliografía
15.
Diagramas
1. Objetivo y campo de aplicación
Este
método especifica el procedimiento de muestreo en fuentes fijas para la
determinación analítica de los siguientes metales: Arsénico (As), Cadmio (Cd),
Cobalto (Co), Cromo (Cr), Cobre (Cu), Estaño (Sn), Manganeso (Mn), Mercurio
(Hg), Níquel (Ni), Selenio (Se), Plomo (Pb), Zinc (Zn).
2. Referencias
Este
método se complementa con las siguientes normas mexicanas vigentes:
2.1 NMX-AA-009-93,
Determinacion del Flujo de gases en un conducto por medio del tubo pitot.
2.2 NMX-AA-054-1978,
CONTAMINACION ATMOSFERICA.- DETERMINACION DEL CONTENIDO DE HUMEDAD EN LOS GASES
QUE FLUYEN POR UN CONDUCTO.- METODO GRAVIMETRICO.
3. Resumen
Una muestra es extraída de la fuente en
forma isocinética. Las partículas se depositan en un filtro y en una sonda,
mientras que los metales en estado gaseoso se absorben en un tren de impactores
que contienen soluciones acuosas diluidas de ácido nítrico y peróxido de
hidrógeno (analizadas para todos los metales incluyendo el mercurio), y
solución ácida de permanganato de potasio (que será utilizada sólo para
análisis de mercurio). Las muestras recuperadas son digeridas y sus fracciones
son analizadas para mercurio (Hg) vía Espectroscopia de Absorción Atómica por
Vapor Frío (EAAVF), y para, As, Cd, Cr, Co, Cu, Hg, Pb, Mn, Ni, Se, Sn y Zn,
por Espectrofotometría de Absorción Atómica (EAA). La Espectroscopia de
Absorción Atómica con Horno de Grafito (EAAHG) puede ser utilizada si se
requiere mayor sensibilidad para el As, Cd, Co, Pb, y Se, se puede utilizar EAA
para todos los metales siempre y cuando se cumplan los niveles de detección de
método expresados a condiciones de ducto planteados como objetivo del protocolo
de prueba. De manera similar, Plasma Acoplado Inducido-Espectrometría de Masas
(ICP-MS).
4. Interferencias
4.1 Desarrollo de flujo ciclónico (En la
sección de control de calidad se describe el procedimiento de evaluación del
grado de desarrollo de flujo (o Flujo Ciclónico).
4.2 Arreglos permisibles entre
Pitot-Boquilla-Sonda-Termopar; en la sección de control de calidad se describe
el procedimiento para verificar los arreglos.
4.3 Interferencias físicas: Humedad,
temperatura, y partículas suspendidas
5. Definiciones
|
5.1
Puerto de Muestreo: |
Perforación y aditamento realizado en el ducto para acceso a su
interior por equipos de muestreo. |
|
|
|
|
5.2
Punto Transversal: |
Ubicación dentro del área transversal de flujo seleccionado para la
extracción de muestra y/o evaluación de velocidad. |
|
|
|
|
5.3
Condiciones estándar: |
Son aquellas en que la temperatura es de 25°C y la presión es de
101325 Pa. |
|
|
|
|
5.4
Condiciones Reales: |
Temperatura y presión a la que se encuentra el gas dentro del ducto
en el sitio de muestreo. También conocidas como "Condiciones
Actuales", "Condiciones de Ducto" o "Condiciones de
Operación". |
|
|
|
|
5.5
Barrida: |
Conjunto de puntos transversales ubicados en una misma dirección
dentro de un corte transversal de flujo. |
|
|
|
|
5.6
Diámetro Equivalente (DE): |
Diámetro interno
equivalente a cuatro veces el radio hidráulico del ducto, o bien, cuatro
veces el área transversal de flujo entre el perímetro húmedo. DE = 4 x rH = 4 AT / PH |
|
|
|
|
5.7
Area Transversal de Flujo (AT): |
Area comprendida por los límites internos del ducto que se ubica en
forma perpendicular a la dirección del flujo gaseoso. |
|
|
|
|
5.8
Perímetro Húmedo (PH): |
Perímetro interno del ducto (el que tiene contacto con el gas
directamente). |
|
|
|
|
5.9
Presión Dinámica (?P): |
Diferencia de presiones (cabezal) formado en un dispositivo de presión
diferencial conectado a un Tubo de Pitot. Dicho cabezal es generado por la
diferencia de presiones de impacto y estática de un gas en movimiento.
También conocida como "Cabezal de Velocidad". |
|
|
|
|
5.10
Presión de Estática (PE): |
Presión generada por la energía interna del gas. También conocida
como "Presión Manométrica" o "Presión Negativa (-)". |
|
|
|
|
5.11
ORSAT |
Equipo utilizado para la determinación de la composición en base
seca de bióxido de carbono (CO2), oxígeno (O2) y en algunos casos monóxido de carbono
(CO). |
|
|
|
|
5.12
Base Seca |
Se refiere a la relación entre un parámetro con respecto a un total
en el que se excluye el contenido de agua (H2O). |
|
|
|
|
5.13
Humedad: |
Es la relación entre el volumen de agua en estado gaseoso existente
en una mezcla de gases, entre el volumen total de la mezcla de gases,
expresada en forma porcentual (% en volumen). |
|
5.14
Contenido de Humedad: |
Igual a Humedad. |
|
5.15
Humedad de Saturación: |
Es la humedad máxima que permite un gas en función al equilibrio
termodinámico existente a una temperatura y presión dada. |
6. Equipo y materiales
6.1
Equipo para muestreo: Tren de muestreo que consta de los siguientes elementos:
(Ver Figura No.1)
Figura
1.- Tren de Muestreo
6.1.1 Boquilla.- Con
punta biselada, construida en vidrio de borosilicato si las temperaturas son
menores a 450ºC; para temperaturas por
encima de 450ºC utilizar boquillas de cuarzo. El ángulo de biselado debe ser 30º, el biselado
debe realizarse en la parte externa para mantener el diámetro interno
constante. El diseño de la boquilla debe ser del tipo de gancho. El rango de
diámetros de boquilla apropiado para muestreos isocinéticos es entre 0,32 a
1,27 cm., e inclusive mayores si el proceso así lo requiere. Es recomendable que
se tenga un juego de boquillas con diámetros internos entre el rango mencionado
anteriormente y con incrementos de 0,16 cm. por lo que el juego de boquillas
comúnmente utilizado consiste de 7 boquillas con diámetros internos de
1/8, 3/16, 1/4, 5/16, 3/8, 7/16 y 1/2 pulgadas. Calibrar las boquillas de
acuerdo a la sección de control de calidad
6.1.2 Tubo interno de la sonda de muestreo
(Liner).
El liner es el tubo por el que la muestra es
extraída, éste forma parte de la sonda de muestreo, debe ser construido en
vidrio de borosilicato o cuarzo, dependiendo de la temperatura de los gases a
extraer del ducto, (para temperaturas menores a 450ºC se puede utilizar vidrio
de borosilicato o cuarzo indistintamente; para temperaturas por encima de 450ºC
usar cuarzo). Si el proceso lo permite durante el muestreo, se deberá mantener
una temperatura de 120 ± 14ºC. Para procesos con altas temperaturas se permite
el uso de liners enfriados en coraza de agua o aire, siempre y cuando el
enfriamiento no provoque condensaciones en la parte frontal del tren de
muestreo (recorrido de boquilla a filtro).
6.1.3 Tubo de Pitot tipo “S”.
El arreglo se presenta en la Figura No. 2,
construido en acero inoxidable, el diámetro externo de la tubería utilizada (Dt)
debe estar entre 0,0048 m y 0,0095 m. Debe existir una distancia igual desde la
base de cada pierna del Tubo de Pitot hasta el plano de la cara abierta
(dimensiones PA y PB), y debe ser
entre 1,05 y 1,50 veces el valor del diámetro externo de la tubería utilizada
(Dt). Las caras abiertas del Tubo de Pitot deberán ser alineadas
como se muestra en la figura No. 3, sin embargo, pequeñas desalineaciones son
permitidas como se muestra en la figura No. 4. El Tubo de Pitot “S” deberá ser fijado a la sonda de muestreo,
como se indica en las figuras 5 y 6, para permitir el monitoreo constante de la
velocidad de los gases. Se deberán cumplir los arreglos de
Pitot-Sonda-Boquilla-Termopar que eliminen el potencial de interferencia en las
mediciones de velocidad.
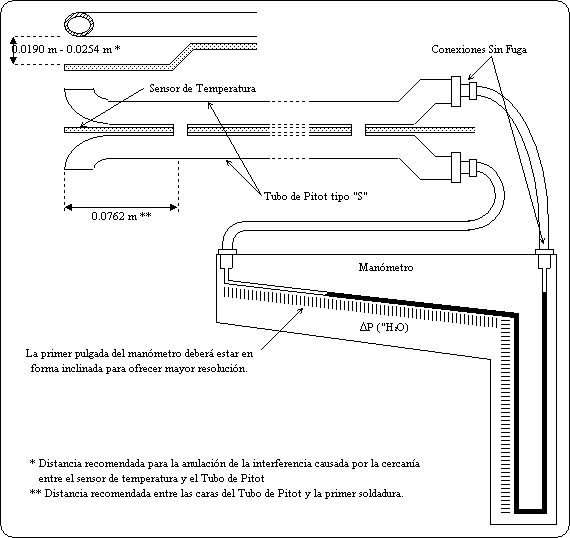
Figura 2
Arreglo Tubo de Pitot tipo "S" -
Manómetro.
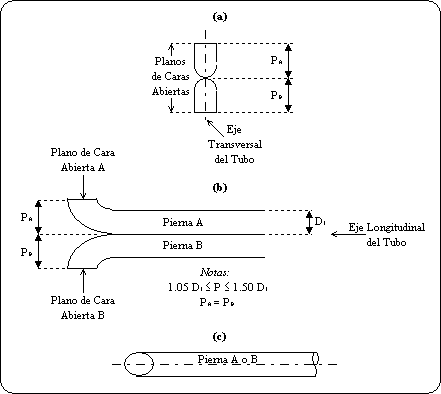
Figura 3
Construcción apropiada de Tubos de Pitot tipo
"S".
(a)
Vista Frontal; planos de caras abiertas perpendiculares a eje transversal del
Tubo.
(b)
Vista Superior; planos de caras abiertas paralelos al eje longitudinal del
Tubo.
(c)
Vista Lateral; ambas piernas de igual distancia y líneas centrales
coincidentes, cuando se observa por ambos lados.
Coeficientes de Calibración Base de 0,4 pueden
ser asignados a Tubos de Pitot tipo "S" construidos de esta manera.
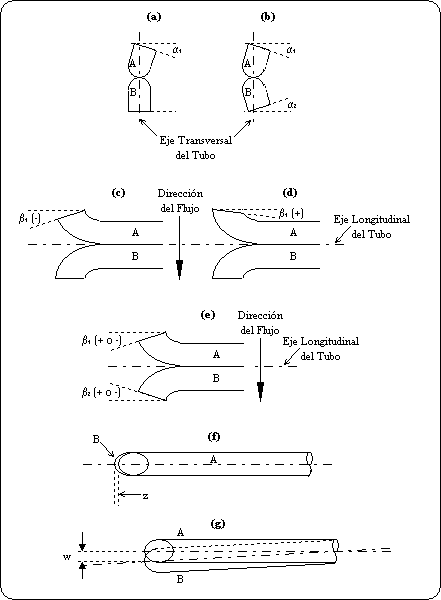
Figura
4.- Tipos de desalineamientos que pueden ocurrir en un Tubo de Pitot tipo
"S", cuando es utilizado en campo o posee una construcción errónea.
Estos no afectarán el Coeficiente de
Calibración Base siempre y cuando: 1 y 2 = 10°, â1 y â 2 = 5°, z = 0.0032 m y w = 0,0008 m.
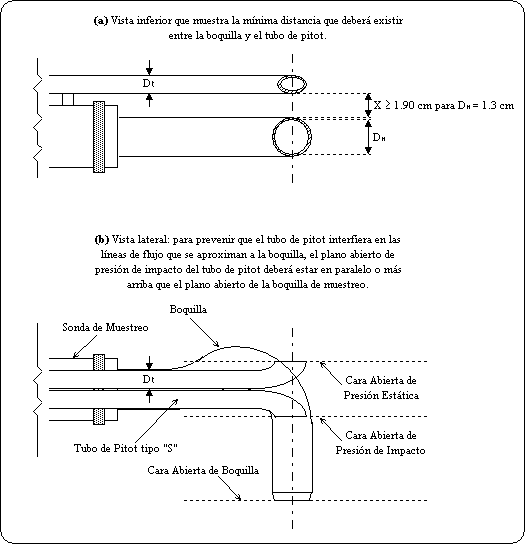
Figura
5.- Configuración del arreglo Tubo de Pitot tipo "S"-Boquilla
requerido para evitar interferencia aerodinámica. Boquillas de tipo gancho. Los
centros de la boquilla y el tubo de pitot tipo "S" deberán ser
alineados. Para Dt entre 0,48 y 0,95 cm
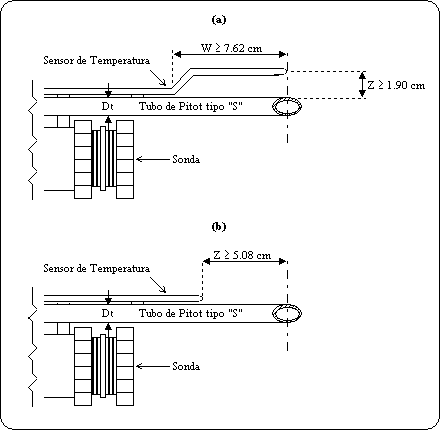
Figura 6.- Ubicación
apropiada del sensor de temperatura para la anulación de posibles
interferencias aerodinámicas y de detección de temperatura.
Las
opciones (a) y (b) pueden ser utilizadas.
Nota. El uso del tubo
Pitot estándar o “L” es útil cuando los niveles de bloqueo en el ducto sean los
suficientemente altos como para separar el tubo Pitot del cuerpo de la sonda.
6.1.4 Manómetro de Presión Diferencial.
Se
requiere de dos manómetros; uno para la determinación de la velocidad y otro
para la determinación de la caída de presión en el medidor del orificio.
6.1.4.1 Se
utiliza un manómetro inclinado-vertical o algún dispositivo análogo. La mayoría
de los equipos de muestreo poseen manómetros de 254 mm de agua con divisiones
de 0,254 mm H2O, en
la sección inclinada con escala de 0 a 25,4 mm H2O y divisiones de 2,54 mm H2O en la sección
vertical con escala de 25,4 a 254 mm H2O, este es adecuado para medir ?P tan baja
como 1,27 mm H2O.
Sólo se permite usar otro tipo de manómetro (o algún otro dispositivo) si
presenta una sensibilidad equivalente o mayor. No obstante se puede utilizar un
medidor de presión diferencial de mayor sensibilidad a criterio del responsable
técnico del muestreo o de la autoridad competente, si se presenta alguna de las
siguientes circunstancias: (1) el promedio aritmético de todos los valores de
?P es menor a 1,27 mm H2O;
(2) para mediciones de 12 o más puntos transversales, en la que más de 10% de
las lecturas individuales de ?P sean menores a 1,27 mm H2O; (3) para
mediciones de menos de 12 puntos transversales, en la que más de una de las
lecturas de ?P sean menores a 1,27 mm H2O
6.1.4.2 Como alternativa a los criterios (1) a (3)
descritos anteriormente, el siguiente procedimiento puede utilizarse para la
determinación si se usa un dispositivo de mayor sensibilidad:
Ecuación
No. 1
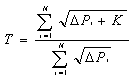
Donde:
T = Parámetro para
Determinación de Aplicabilidad de Dispositivo de Presión Diferencial.
?Pi = Lectura
de Presión Dinámica Individual del Punto Transversal "i", mm H2O
K = 0,127 mm H2O
cuando se utilizan unidades métricas
N = Número Total de Puntos
Transversales
Si T es mayor a 1,05 los datos de presión
dinámica serán inaceptables y se requiere de un dispositivo de mayor
sensibilidad.
6.1.4.3 Si
se utiliza algún manómetro diferencial que no sea del tipo de columna inclinada
(por ejemplo, manómetros magnagélicos), éstos deberán ser verificados en su
calibración antes de cada prueba. Como se indica en la sección de control de
calidad
6.1.5 Portafiltro.
El
portafiltro debe ser de vidrio de borosilicato o cuarzo con soporte de fibra de
vidrio poroso, PTFE o cuarzo y empaque de hule de silicón con recubrimiento de
PTFE. El portafiltro debe estar diseñado en forma tal que garantice su
hermeticidad. El portafiltro debe unirse herméticamente al liner (o al ciclón
en caso de usarse).
6.1.6 Sistema de Calentamiento del Filtro o Caja Caliente
Sistema de calentamiento capaz de mantener al
filtro a una temperatura de 120 ± 14ºC. Debe incluirse algún sensor de
temperatura capaz de medir la temperatura alrededor del filtro con una
precisión de ± 3ºC, con el objetivo de controlar y monitorear la temperatura en
el filtro.
6.1.7 Caja Fría o Condensador.
6.1.7.1 Se utiliza para determinar el contenido de humedad en el gas
muestreado, así como para capturar selectivamente a los metales encontrados en
fase gaseosa. Puede consistir de 4 a 7 impactores conectados en serie, usando
conexiones no contaminantes (se recomienda el uso de unión con rosca y/o sello
con empaque de PTFE). El arreglo de impactores es el siguiente:
Tabla 1.
Secuencia de
Impactores en Caja Fría
|
Impactor |
Uso |
Tipo
Greenburg Smith con Punta |
Objetivo |
Contenido |
|
1 |
Opcional* |
Modificada |
Eliminar Condensados |
Vacío |
|
2 |
Obligatorio |
Modificada |
Captura por Reacción Química de Todos los
Metales excepto parte del Mercurio |
Solución de HNO3 / H2O2 |
|
3 |
Obligatorio |
Estándar |
Captura por Reacción Química de Todos los
Metales excepto parte del Mercurio |
Solución de HNO3 / H2O2 |
|
4 |
Opcional** |
Modificada |
Eliminar arrastre de la disolución de HNO3 / H2O2 a la disolución
de H2SO4 / KMnO4 |
Vacío |
|
5 |
Opcional** |
Modificada |
Captura por Reacción Química de Mercurio |
Solución de H2SO4 / KMnO4 |
|
6 |
Opcional** |
Modificada |
Captura por Reacción Química de Mercurio |
Solución de H2SO4 / KMnO4 |
|
7 |
Obligatorio |
Modificada |
Expulsar el gas |
Sílica Gel |
* Se recomienda utilizar cuando la corriente
gaseosa posee un alto contenido de humedad.
** Se pueden eliminar cuando no se requiere la
medición de mercurio
6.1.7.2 Usar
conexiones flexibles de PTFE entre impactores, portafiltros y a la entrada del
sistema de impactores, siempre y cuando la temperatura de los gases lo permita
(la temperatura máxima recomendada para el PTFE es de 200ºC).
6.1.7.3 Instalar
un termómetro con subdivisiones de 1ºC en el último impactor.
6.1.7.4 El sistema de impactores debe sumergirse en
un baño de agua y hielo. La temperatura en la salida del último impactor debe
monitorearse y mantenerse por debajo de 20ºC.
6.1.8 Caja de Control o Sistema de Medición.
Consta de un vacuómetro, bomba de vacío,
sensores de temperatura con una sensibilidad de ± 3ºC, gasómetro seco capaz de
medir dentro de 2%, medidor de orificio y equipo adicional (ver Figura No.1 del tren de muestreo). Se puede
utilizar algún otro sistema de medición capaz de mantener velocidades de
muestreo dentro de ± 10% de condiciones isocinéticas, y de determinar el
volumen de muestra dentro de un 2%.
6.1.9
Equipo para Determinación de la Densidad del Gas.
6.1.9.1 Debe
incluir un sensor de temperatura y manómetro de presión (descritos
anteriormente). Analizador de composición de gas capaz de determinar los
principales componentes de la mezcla gaseosa seca (generalmente CO2, O2, CO y N2). Se puede
utilizar analizadores instrumentales de cada compuesto. El nitrógeno (N2) es determinado
por diferencia e incluye el resto de los compuestos que se encuentran en traza.
6.1.9.2 El
sensor de temperatura deberá estar fijo a la sonda de muestro y/o tubo de
pitot, en algún arreglo en que la punta del sensor sobrepase el cobertor de la
sonda y que no toque ningún metal. En caso de que la sonda posea un tubo de
Pitot, el sensor deberá ser fijado de acuerdo a alguno de los dos arreglos
descritos en las figuras de Tubos de Pitot anteriores.
6.1.10
Balanza Granataria.- Con una precisión de 0,5 g
6.2 Material para recuperación de muestras
6.2.1 Escobilla.-
Construida en PTFE y cerdas de nylon
6.2.2 Frascos para recuperación de los lavados.-
Frascos de vidrio con tapa de cierre hermético con contratapa cubierta de PTFE,
capacidad de 500 a 1,000 ml
6.2.3 Caja Petri de polietileno o vidrio, de un tamaño
adecuado para contener los filtros de las muestras sin necesidad de ser
doblados.
6.2.4
Probeta Graduada.- Con subdivisiones menores o igual a 2,0 ml
6.2.5
Filtros que no posean aglomerados orgánicos. Estos filtros deben contener menos
de 1,3 in2 de cualquier
metal a analizar. Los filtros deben ser de cuarzo.
6.2.6
Embudos para filtración de polietileno
6.3
Material para preparación de disoluciones que capturan los metales en el
muestreo
6.3.1
Matraz volumétrico de 1,000 ml
6.3.2
Probeta Graduada de 100 ml
6.3.3
Pipeta volumétrica de 1,0 ml
6.3.4
Pipeta graduada de 10 ml
6.3.5
Vidrio de reloj
6.3.6
Embudo de vidrio para filtración
6.3.7
Frascos reactivo para almacenar la disolución de permanganato de potasio ácido,
capacidad de 1,000 ml
7.
Reactivos y disoluciones
7.1
Reactivos utilizados en el lavado de material para el muestreo:
7.1.1
Acido Nítrico (HNO3)
concentrado grado reactivo
7.1.2 Agua
destilada libre de metales Tipo II ASTM
7.1.3
Detergente libre de compuestos metálicos
7.2
Reactivos para la preparación de disoluciones utilizadas en el muestreo
7.2.1 Agua destilada tipo ASTM II
7.2.2 Acido nítrico
concentrado grado absorción atómica
7.2.3 Acido clorhídrico
concentrado grado absorción atómica
7.2.4 Peróxido de
hidrógeno al 30% grado reactivo analítico (ACS)
7.2.5 Acido sulfúrico concentrado grado reactivo
analítico (ACS)
7.2.6 Permanganato de potasio grado reactivo
analítico (ACS)
7.2.7 Acetona grado reactivo analítico (ACS)
concentración de residuos sólidos menor o igual a 0,001% en peso.
7.3 Preparación de
disoluciones
7.3.1 Disolución para
captura de metales de ácido nítrico (HNO3) y peróxido de hidrógeno (H2O2), 5% HNO3 / 10% H2O2.- Adicionar 50 ml de HNO3 concentrado a un
matraz volumétrico de 1,000 ml que contenga aproximadamente 500 ml de agua (con
agitación moderada y constante), posteriormente agregar 333 ml de H2O2 al 30%. Aforar al
volumen del matraz con agua.
7.3.2 Disolución de
captura de permanganato de potasio ácido (KMnO4), 4% KMnO4 (P/V), 10% H2SO4 (V/V).- Esta disolución debe ser preparada el
día del muestreo. Adicionar agitando cuidadosamente 100 ml de ácido sulfúrico
concentrado a 800 ml de agua en un matraz volumétrico de 1,000 ml. Aforar al
volumen del matraz con agua. En otro matraz volumétrico de 1,000 ml, disolver
40 g de KMnO4 en
solución de H2SO4 al 10%, aforar al
volumen del matraz con disolución de H2SO4 al 10%. Esta disolución es la denominada
captadora de permanganato de potasio ácido.
NOTA: Para prever una
de descomposición autocatalítica de la disolución de permanganato, filtrarla a
través de un filtro número 541.
También, debido a la reacción entre el permanganato de potasio y el ácido, se
puede incrementar la presión en el frasco de almacenado, por lo que se
recomienda no llenar totalmente los recipientes y dejarlos ventear para liberar
la presión. El venteo se requiere, sin embargo se recomienda que éste sea
mínimo para evitar contaminación.
7.3.3 Disolución de
ácido nítrico (HNO3)
0,1 N.- Agregar aproximadamente 6,3 ml de HNO3 concentrado (70%)
a un matraz volumétrico de 1,000 ml que contenga aprox. 900 ml de agua. Aforar
con agua al volumen del matraz.
7.3.4 Acido clorhídrico
(HCl) 8,0 N.- Agitando, agregar cuidadosamente 690 ml de HCl concentrado a un
matraz volumétrico de 1,000 ml que contenga aprox. 250 ml de agua. Aforar con
agua al volumen del matraz.
7.3.5 Acido nítrico
(HNO3)
al 10% (V/V).- Esta disolución se usa para lavar el material.- Agitando,
agregar 500 ml de HNO3
concentrado a un recipiente que contenga aproximadamente 4,000 ml de agua,
completar a un volumen de aproximadamente 5,000 ml con agua.
8. Preservación y almacenamiento de muestras
8.1 Colectar un
volumen de muestra homogéneo y representativo superior a 1 L en un frasco de
polietileno o vidrio con tapa de boca ancha, teniendo siempre en cuenta que el
material en suspensión no debe adherirse a las paredes del recipiente.
8.2 No se recomienda la adición de agentes
conservadores. Transportar la muestra y mantenerla a 4°C hasta realizar el
análisis. Las muestras deben estar a temperatura ambiente al momento del
análisis.
9. El tiempo máximo de almacenamiento previo al análisis
es de 7 días. Sin embargo, se recomienda realizar el análisis dentro de las 24
horas posteriores a su colecta.
9.
Control de calidad
9.1 Criterio para la
Determinación de Ausencia de Flujo Ciclónico.
9.1.1 En ángulo nulo
absoluto () promedio
obtenido de todos los puntos transversales, deberá ser menor o igual a 20°,
para poder establecer que no existe flujo ciclónico, y por lo tanto, considerar
adecuado el sitio de muestreo propuesto, de lo contrario, deberá procederse a:
(1) cambiar la ubicación del sitio de muestreo, o (2) utilizar algún mecanismo antes del sitio de muestreo el cual
tenga la propiedad de alinear el flujo.
9.1.2 Verificación de
la Ausencia de Flujo Ciclónico.
9.1.2.1 En la mayoría de
los ductos, la dirección del vector principal de velocidad (líneas de flujo) se
encuentra en forma paralela a las paredes del ducto, motivo por el cual, la
referencia para colocar los dispositivos para la determinación de velocidad
(Tubo de Pitot), es la pared del ducto. Sin embargo, un patrón de flujo
ciclónico puede presentarse en varios casos: (1) después de colectores
ciclónicos y deshumidificadores inerciales seguidos de lavadores vénturi, o (2)
en ductos que tengan entradas tangenciales u otras configuraciones que generen
flujos circulares. En estos casos, los vectores principales de velocidad
(líneas de flujo), pueden no estar paralelos a la pared, y por lo tanto, la
pared no funciona como referencia para el alineamiento de dispositivos de
medición de flujo. También, existen casos en los que turbulencias con patrón
ciclónico en ciertas secciones del ducto (como son las zonas cercanas a codos,
reducciones, expansiones, abanicos, sopladores, etc.), pueden estar presentes,
por lo que la presencia o ausencia de flujo ciclónico deberá ser evaluada con
el objetivo de determinar si el sitio de muestreo seleccionado es el adecuado.
9.1.2.2.
Determinación del Angulo Nulo.
Para la verificación de la presencia de flujo
ciclónico, se requiere del uso de un Tubo de Pitot tipo "S" descrito
más adelante, y un transportador de ángulo. Nivelar y ajustar en cero un
manómetro de agua inclinado. Colocar el Tubo de Pitot tipo "S" en
cada uno de los puntos transversales de manera que las caras abiertas del tubo
queden en forma paralela a la pared del ducto (referencia para la dirección del
flujo). Ajustar un transportador de ángulo al cuerpo del Pitot de manera que
quede indicando un ángulo de 0°, denominándose como "Posición de
Referencia 0°", la cual implica que si las líneas de flujo del gas en cada
punto transversal son paralelas a la referencia de dirección de flujo (pared),
la lectura en el manómetro deberá indicar 0 "H2O, debido a que
ambas caras registrarán únicamente la presión estática del gas en ese punto,
las cuales se contrarrestarán para indicar una caída de presión nula en el
manómetro. De lo contrario, si el flujo se mueve con alguna diferencia de
ángulo con respecto a la pared, el manómetro indicará alguna caída de presión
(positiva o negativa). Rotar el Pitot hacia cualquiera de los dos lados de
manera que se logre obtener una caída de presión nula en el manómetro. Al
lograrse, leer el ángulo resultante en el transportador, y registrarlo como el
ángulo nulo () (en forma
absoluta y con resolución de hasta 1°), de dicho punto transversal. El ángulo
nulo se define como el ángulo en el que no se registra alguna resultante del
vector principal de velocidad (ver figura del ángulo nulo), y por ende, el
ángulo de flujo con respecto a la referencia (pared). Es importante aclarar que
el responsable técnico de muestreo deberá: (1) tomar como referencia las
pruebas y procedimientos requeridos para el control de calidad de resultados en
el manejo de sistemas de Tubo de Pitot-Manómetro, y (2) no realizar la prueba
intentando medir el ángulo en el que se presenta la mayor caída de presión
(ángulo ß), debido al error existente por causa de interpretación de
resultados.
Figura del Angulo
Nulo
9.1.2.3
Verificación de calibración para cada boquilla del juego, determinar en un
mínimo de tres distintos diámetros de la boquilla, y calcular su promedio. De
las tres mediciones, la diferencia entre la máxima y la mínima no podrá exceder
de 0,1 mm. En caso de excederse esta diferencia, la boquilla deberá corregirse
o substituirse
9.1.3
Calibración del Tubo de Pitot tipo "S".- Deberá poseer un coeficiente
de calibración conocido, determinado ya sea por Calibración Geométrica para la
obtención de un “Coeficiente de Calibración Base”, o bien mediante una
Calibración Experimental en Base a Túnel de Viento para la obtención de un
“Coeficiente de Calibración Experimental”. Se deberá identificar cada uno de
los tubos Pitot con una grabación en la parte externa permanente
9.1.3.1 Un
Tubo de Pitot Estándar podrá ser utilizado en lugar de un tipo "S",
siempre y cuando cumpla con lo establecido, sin embargo, las caras abiertas
para presión estática y de impacto pueden ser tapadas por las partículas que
vayan suspendidas en el gas. Por lo tanto, cuando un Tubo de Pitot Estándar sea
utilizado para realizar una evaluación, se deberán presentar pruebas adecuadas
de que las aberturas no fueron bloqueadas durante el muestreo, lo cual deberá
hacerse según el siguiente procedimiento:
(1) Una vez terminada una barrida de lecturas deberá sacarse
el Tubo de Pitot Estándar y limpiarse con aire a presión en dirección hacia
afuera de las aperturas;
(2) Colocarlo de nuevo en el primer punto
transversal evaluado y registrar la ?P
obtenida como ?PL (se podrá efectuar esta prueba
en todos los puntos transversales o únicamente en el primero o el último);
(3) La ?P obtenida antes y después de la limpieza (?P y ?PL
respectivamente) no deberá variar por más de un 5% absoluto, utilizando la
siguiente ecuación:
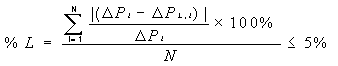
Donde:
%L = % de Variación ?P antes y después de Limpieza de Tubo de Pitot.
?Pi = Presión Dinámica en Punto
Transversal "i".
?PL,i = Presión Dinámica Después de
Limpieza en Punto Transversal "i".
N = Número de Puntos Transversales.
(4) El operador deberá tener cuidado con variaciones en el proceso
que indiquen cambios en la velocidad del gas, lo cual puede llegar a traer un
cambio en la ?P obtenida antes y después de
la limpieza, el cual no hubiese sido causado por algún bloqueamiento de
las aperturas en el tubo, y también,
cuando el proceso evaluado registre una frecuencia de variación de velocidades
muy corta, la cual se observa en un pequeño movimiento oscilatorio en el
manómetro, el operador deberá de registrar el valor promedio, mínimo y máximo
del rango de oscilación antes y después de la limpieza, para poder concluir si
se bloqueó alguna de las aperturas durante el muestreo. Control de calidad
9.1.4 Para
verificar la calibración del manómetro diferencial se deberá comparar las
lecturas de presión dinámica del dispositivo contra las lecturas de un
manómetro de columna de aceite en un mínimo de 3 puntos (los cuales representen
el rango promedio de presiones dinámicas en el ducto). Si en cada punto, los
valores de ?P de ambos instrumentos concuerdan dentro de un 5%, se considerará
que el dispositivo calibrado es adecuado, de lo contrario, se deberá evitar el
uso de este manómetro o proceder a ajustar el manómetro para una calibración
adecuada.
9.1.5
Sensibilidad:
La
siguiente Tabla muestra los niveles de detección típicos de este método
utilizando EAAHG e ICAP:
|
Metal |
Nivel
de Detección (g/m3 C.N.B.S.) |
|
|
|
ICAP |
EAHG |
|
Arsénico |
19.43 |
0.41 |
|
Cadmio |
1.53 |
0.03 |
|
Cobalto |
2.54 |
0.31 |
|
Cobre |
2.14
(a) |
0.31 |
|
Cromo |
2.54 |
|
|
Estaño |
|
|
|
Manganeso |
0.71 |
0.31 |
|
Mercurio |
0.57
(b) |
|
|
Níquel |
5.49
(a) |
|
|
Plomo |
15.36 |
0.81 |
|
Selenio |
27.46 |
|
|
Zinc |
0.81
(a) |
1.02 |
Tabla: Nivel
Mínimo de Detección
(microgramos por
metro cúbico normal base seca).
(a) Análisis por AAS.
(b) Análisis por CVAAS.
Si se requiere, los niveles de detección
pueden ser menores a los mostrados, siguiendo las siguientes opciones:
· Un muestreo de 1 hora puede obtener una
muestra de gas seco de 1,25 m³ a condiciones normales. Si el tiempo de muestreo
es aumentado y el volumen de gas seco muestreado asciende a 5 m³ a condiciones
normales (4 veces mayor), el nivel de detección sería un cuarta parte el
mostrado en la Tabla. Lo anterior es aplicable para muestreos aún más largos
(la relación en que se aumenta el volumen de muestra, es la relación en que se
divide el nivel de detección).
· El nivel de detección asume que toda la
muestra es digerida (con la excepción de la alícuota de mercurio) y los
volúmenes finales de muestra para análisis son 300 ml para la parte frontal del
tren de muestreo y 150 ml, Fracción 2A, para la parte trasera del tren de
muestreo. Si el volumen de muestra de la parte frontal y trasera es reducido,
el nivel de detección será reducido en forma proporcional.
· Cuando los dos anteriores puntos son
efectuados al mismo tiempo, los efectos son multiplicativos. Por ejemplo, si el volumen de gas seco
muestreado es incrementado a razón de un factor de 5, y el volumen de muestra
para análisis es disminuido a razón de un factor de 6, entonces el nivel de
detección mostrado en la Tabla es reducido a razón de un factor de 30.
· Contrariamente, reducir el volumen de gas
seco muestreado y aumentar el volumen de muestra líquida para análisis, aumenta
el límite de detección mostrado en la Tabla (por lo tanto el método es menos
sensible).
· La discusión anterior asume que no hay
corrección por blanco.
9.1.6
Precisión
La precisión para cada metal (desviación
estándar relativa) del método es mostrada en la siguiente Tabla:
|
Metal |
Precisión (%) |
|
As |
13.5 |
|
Cd |
11.5 |
|
Cr |
11.2 |
|
Cu |
11.5 |
|
Pb |
11.6 |
|
Sn |
|
|
Se |
15.3 |
|
Zn |
11.8 |
|
Ni |
7.7 |
|
Mn |
No Determinado (a) |
|
Hg |
No Determinado (a) |
Tabla: Precisión
por Metal
(a)
Se sospecha un valor semejante al resto de los metales.
9.1.7 Es
común que se utilice el principio instrumental de polarografía (celdas
electroquímicas), las cuales no determinan el CO2 vía instrumental, sino que lo calculan de
acuerdo a la función del tiempo de combustible quemado y al contenido de
oxígeno que se encuentra analizando, sin embargo;
a) para
procesos que no queman combustible fósil; b) en que la composición del
combustible varía constantemente; c) en que no se utiliza aire o se enriquece
con mezclas de O2/N2 distintas a las
del aire o d) que se genere bióxido de carbono por procesos distintos a la
combustión, el cálculo se deberá efectuar vía factor de combustible
(equivalente a un balance de materia simplificado) y la determinación debe ser
por vía experimental (ORSITE; FYRITE o
instrumental para CO2).
9.2
Aspectos generales de control de calidad
9.2.1 Cada
laboratorio que utilice este método está obligado a operar un programa de
control de calidad (CC) formal. Los requerimientos mínimos de este programa
consisten en una demostración inicial de la capacidad del laboratorio para
cumplir con las especificaciones de desempeño del método, además realizar
análisis continuos de muestras de control de calidad (MCC) para demostrar la
precisión y exactitud continuas y el análisis de blancos. El desempeño del
laboratorio debe compararse con los criterios aquí establecidos, con objeto de
determinar si los resultados de los análisis cumplen con las especificaciones
de desempeño del método. El analista debe hacer una demostración inicial de su
habilidad para generar una exactitud y precisión aceptables por este método.
Esta habilidad debe realizarse como se menciona en la Sección de desempeño del
método.
9.2.2 Cada
vez que se realice una modificación al método o que se cambie el analista
responsable de llevar a cabo esta determinación, el analista designado debe
repetir el procedimiento mencionado en la sección 9.2, si el cambio va a afectar el límite de detección del método
(LDM), el laboratorio debe demostrar que el nuevo LDM determinado es igual o
más bajo que el anterior para los analitos de interés.
9.2.3 No
se permite el uso de técnicas determinativas alternativas y cambios que
degraden la ejecución del método. Si se utiliza una técnica analítica que no
sea la especificada en este método, dicha técnica debe tener especificaciones
iguales o mejores que las de la técnica descrita en este documento para el
analito de interés.
9.2.4 Es
obligatorio para el laboratorio mantener los registros de las modificaciones
hechas a este método. Estos registros deben de incluir lo siguiente:
- La justificación por escrito de la necesidad
de realizar modificaciones al método para ese analito.
- Los nombres, títulos, direcciones y número
de teléfono de los analistas que ejecutaron los análisis y modificaciones y el
encargado de control de calidad que presenció y verificó los análisis y sus
modificaciones.
- Los resultados de todas las pruebas de
control de calidad del método modificado comparadas con el método original,
dichos datos deben de incluir todos los parámetros mencionados en la Sección de
capacidad inicial del método.
- La información escrita en las bitácoras
tanto del equipo como del analista, deben incluir los siguientes datos:
a) Identificación de la muestra
b) Número del lote analítico en el cual se analizó la
muestra
c) Fecha del análisis
d) Procedimiento cronológico utilizado
e) Cantidad de muestra utilizada
f) Número de muestras de control de calidad analizadas en
el lote
g) Trazabilidad de las calibraciones de los
instrumentos de medición
h) Registros de bitácoras, en cintas de respaldo o en otros
respaldos de información
i) Información cruda reportada por los equipos o por los
analistas
j) Evidencia de la aceptación o rechazo de los resultados
del lote analítico.
9.3 En
cada muestreo definitivo de metales se debe realizar de manera simultánea un
análisis de peso molecular promedio vía continua y/o integral (el cual incluye
el contenido de oxígeno). Dadas las variaciones de contenido de oxígeno entre
cada muestreo, pueden ser sustanciales y generar errores significativos por
corrección de concentraciones a niveles de referencia de oxígeno (7%). Calcular
la concentración de oxígeno en cada corrida para considerarlo en el cálculo de
la corrección por dilución.
10.
Procedimiento
10.1
Determinaciones preliminares
10.1.1 Peso
Molecular Promedio Base Seca (MBS)
10.1.1.1 Para
la ejecución de un estudio isocinético, es necesario contar inicialmente con un
estimado del peso molecular promedio base seca de la mezcla gaseosa, el cual se
utiliza inicialmente para la estimación de la constante de proporcionalidad
isocinética. Este peso molecular puede ser estimado en base a criterio o vía
determinación experimental. En caso de que el MBS sea estimado en base a criterio (únicamente
permitido para cálculo preliminar de diámetro de boquilla y Constante de
Proporcionalidad Isocinética), éste deberá ser determinado vía experimental,
durante o al final del estudio isocinético, con el fin de ser utilizado para
cálculo de resultados.
Nota. Es
muy importante tener en cuenta que si la estimación inicial se realiza a
criterio y se comete un error considerable en dicha estimación, la constante de
proporcionalidad isocinética utilizada en el estudio puede quedar lo
suficientemente errónea como para que el estudio sea rechazado por falta de
condiciones isocinéticas de muestreo, por lo que se recomienda que en caso de
tenerse fuerte duda al efectuar la estimación, es preferible efectuar una
determinación experimental.
10.1.1.2 En cada muestreo definitivo de metales se
debe realizar de manera simultánea un análisis de peso molecular promedio vía
continua y/o integral (el cual incluye el contenido de oxígeno). Dado que las
variaciones de contenido de oxígeno entre cada muestreo, pueden ser
substanciales y generar errores significativos por corrección de
concentraciones a niveles de referencia de oxígeno.
10.1.2 Selección del Sitio de Muestreo y Mínimo Número de
Puntos Transversales de Muestreo.
10.1.2.1
Seleccionar el sitio de muestreo y número mínimo de puntos transversales de
muestreo de acuerdo al Método NMX-AA-009-1993 o algún otro Método equivalente.
10.1.2.2 Es
importante mencionar que el número de puntos de muestreo determinados en
el muestreo debe ser mayor o igual al
mínimo especificado en el Método NMX-AA-009-1993 o algún otro método equivalente. También, existen casos
en los que el Método NMX-AA-009-1993 o algún otro método equivalente, habilita
a algún ducto que no posee la geometría adecuada pero que sí específica el
régimen y desarrollo de flujo adecuado,
a que el estudio pueda ser efectuado independientemente de que no se cumplan
los requisitos geométricos establecidos, siempre y cuando los interesados por
el resultado lo aprueben (entidades gubernamentales, ingeniería de procesos,
etc.). Para esto es indispensable que se efectúe la evaluación del nivel de
desarrollo de flujo, o bien, demostrar la ausencia de flujo ciclónico en el
sitio de muestreo.
10.1.2.3
Seleccionar la longitud de sonda y liner adecuado para lograr los puntos de
muestreo determinados
10.1.3
Presión Dinámica, Presión Estática, Presión Absoluta y Temperatura.
10.1.3.1
Determinar la presión dinámica (?P), presión estática (PE) y temperatura
del gas (TC),
conforme a lo indicado en el Método NMX-AA-009-1993 o algún otro método
equivalente, en cada uno de los puntos transversales de muestreo, y calcular
las condiciones promedio conforme a las siguientes ecuaciones:
Ecuación No. 2 y No. 3
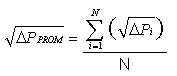
![]()
Donde:
?PPROM = Presión Dinámica Promedio.
?Pi = Presión Dinámica en Punto Transversal "i".
N = Número
Total de Puntos Transversales.
Ecuación
No. 4
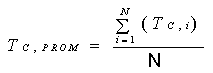
Donde:
TC,
PROM = Temperatura del Gas Promedio.
TC,
i = Temperatura del Gas en Punto Transversal "i".
Ecuación No. 5
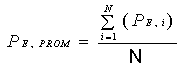
Donde:
PE, PROM = Presión Estática del Gas Promedio.
PE, i = Presión Estática del Gas en Punto
Transversal "i".
Ecuación
No. 6
![]()
Donde:
PC,PROM = Presión Absoluta del Gas Promedio.
PB = Presión Barométrica.
10.1.4
Contenido de Humedad en el Gas (%H).
Este método incluye la determinación
experimental del contenido de humedad en forma simultánea a la determinación de
metales, sin embargo, para el cálculo de la constante de proporcionalidad
isocinética, la cual se requiere previa al muestreo, es necesario conocer con
cierta precisión el por ciento de humedad (%H). Por lo tanto, existe para su cálculo el Método NMX-AA-054-1978 o
algún otro Método equivalente.
10.1.5
Selección del Diámetro de Boquilla.
10.1.5.1
Calcular el diámetro de boquilla requerido para operar a aproximadamente una
caída de presión en el medidor de orificio equivalente a ?H@ (flujo de 0,75 ft3/min a condiciones
de gasómetro), con el uso de la siguiente ecuación:
Ecuación
No. 7
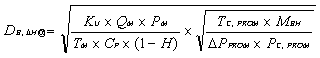
Donde:
DB,DH@ = Diámetro de boquilla requerido para realizar un muestreo con una
caída de presión en el medidor de orificio promedio de aproximadamente ?H = ?H@
lo cual es equivalente a un flujo en el medidor de orificio de 0,75 ft³/min
a condiciones de gasómetro, (“)
QM = Flujo en el medidor de orificio, (0,75 ft³/min)
PM = Presión Absoluta en el medidor de orificio (equivalente a presión
de Gasómetro, PG), (“Hg)
TM = Temperatura Absoluta en el Medidor de Orificio (equivalente a temperatura
de salida de gasómetro), (ºR)
CP = Coeficiente de Tubo de Pitot, (adimensional)
H = Contenido
de Humedad en Fracción Volumétrica, (adimensional)
TC,PROM = Temperatura Absoluta Promedio del Gas en el Ducto, (ºR)
MBH = Peso Molecular Promedio Base Húmeda de la mezcla de gases,
(lb./lbmol)
?PPROM = Presión Dinámica Promedio, (“H2O)
PC,PROM = Presión Absoluta Promedio del Gas en el Ducto, (“Hg)
KU = Constante Dimensional, (0,0357)
10.1.5.2 Una
vez obtenido DB, ?H@, éste
se deberá ubicar dentro de los dos valores de diámetro nominal de boquilla (DBN) más aproximados,
asignando preferiblemente el valor que se encuentre más cercano.
10.1.5.3
Tomar la boquilla seleccionada y asignar el diámetro de boquilla real indicado
en la calibración correspondiente de dicha boquilla, al valor de DB.
10.1.6 Cálculo de la
Constante de Proporcionalidad Isocinética.
La
constante de proporcionalidad isocinética se define como:
Ecuación
No. 8
![]()
Donde:
K = Constante de Proporcionalidad Isocinética
?H = Caída
de Presión en Medidor de Orificio
?P = Presión
Dinámica
Constante
de Proporcionalidad Isocinética con relación de temperaturas constante (K):
Ecuación
No. 9
![]()
Donde:
K = Constante de Proporcionalidad Isocinética
con relación de temperaturas constante.
KK = Constante Dimensional, (849,8)
CP = Coeficiente de Calibración de Tubo de Pitot, (adimensional).
?H@ = Coeficiente de Calibración de Medidor de
Orificio, (“H2O)
TC,PROM = Temperatura Absoluta Promedio de Gases en Ducto
PC,PROM = Presión Absoluta Promedio de Gases en Ducto
MBS = Peso Molecular Promedio Base Seca
MBH = Peso Molecular Promedio Base Húmeda
TM = Temperatura Promedio en el Medidor de Orificio
DB = Diámetro de Boquilla, (pulgadas)
H = Contenido de Humedad en forma de Fracción,
(adimensional)
Nota.
Ambas temperaturas en la ecuación de K deberán manejarse con la misma unidad y
en escala absoluta.
Ambas presiones en la ecuación de K deberán
manejarse con la misma unidad y en escala absoluta.
Ambos pesos moleculares en la ecuación de K
deberán manejarse con la misma unidad.
10.2
Muestreo
10.2.1
Preparación de la pre-prueba
10.2.1.1
Enjuagar todo el material de vidrio del tren de muestreo con agua caliente,
posteriormente lavar con agua caliente jabonosa, después enjuagar tres veces
con agua corriente, seguido de tres enjuagues adicionales.
10.2.1.2
Remojar todo el material de vidrio en una solución de ácido nítrico al 10% v/v,
por un mínimo de cuatro horas, enjuagar tres veces con agua corriente, por
último enjuagar con acetona, permitir que se seque perfectamente.
10.2.2
Preparación del tren de muestreo
10.2.2.1
Colocar el tren de muestreo como en la prueba normal de muestreo isocinético de
partículas suspendidas. Utilizar el primer impactor como una trampa de humedad,
el segundo y el tercer impactor deben contener una solución de ácido
nítrico/peróxido de hidrógeno (HNO3/H2O2), el cuarto impactor deberá permanecer vacío,
el quinto y el sexto deben contener una solución ácida de permanganato de
potasio (KMnO4). El
último impactor debe contener de 200 a 300 g de sílica gel. Alternativamente la
sílica gel puede pasarse directamente en el impactor justo antes del ensamble
final del tren.
10.2.2.2 Basado en las
condiciones específicas de la fuente de muestreo, el uso de un primer impactor
vacío se puede eliminar si la humedad que será colectada en los impactores es
menor a 100 ml. Si no se ha analiza mercurio los impactores 4, 5 y 6 no se
requieren.
10.2.2.3 Para asegurar que
las conexiones del tren de muestreo se encuentren libres de fugas y para
prevenir posibles problemas de contaminación de la muestra, usar cinta de PTFE
u otro material inerte (no usar silicón o grasa).
Precaución.-
Extremar el cuidado para evitar la contaminación dentro del tren, prevenir la
acidificación de peróxido de hidrógeno (H2O2) a causa de la mezcla con permanganato de
potasio KMnO4 en
la acidificación.
10.2.3 Operación del
tren de muestreo
10.2.3.1 Durante la
corrida de muestreo, mantener una tasa de muestreo isocinético dentro del
10% de isocinetismo real a menos que se
indique otra especificación y una temperatura alrededor del filtro de 120 14ºC.
10.2.3.2 Para cada corrida
registrar los siguientes datos; a)
lectura inicial del gasómetro; b)
las lecturas del gas seco al inicio y al final de cada incremento de tiempo en
el muestreo; c) cuando se cambie la
tasa del flujo; d) antes y después
de cada verificación de fugas y; e)
cuando se suspenda el muestreo.
10.2.3.3 Para disminuir la
contaminación cruzada con el material depositado durante el muestreo, limpiar
el puerto antes de iniciar la prueba. Para el muestreo remover la tapa de la
boquilla, verificar que el filtro y el sistema de calentamiento de la sonda
están incrementando la temperatura, y que el tubo Pitot y la sonda están
colocados correctamente. Colocar la boquilla en el primer punto transverso con
la extremidad apuntando directamente dentro del vapor de gas. Inmediatamente
iniciar el bombeo y ajustar el flujo a condiciones isocinéticas.
10.3 Recuperación de
la muestra
10.3.1 Iniciar los
procedimientos de limpieza tan pronto como sea posible, después que la sonda
sea retirada de la chimenea al final del periodo de muestreo.
10.3.1.1 Permitir que la
sonda se enfríe antes de recuperar la muestra. Cuando se pueda manejar de forma
segura, limpiar todas las partículas del exterior, cerca de la punta de la
boquilla de la sonda y colocar un tapón, previamente lavado y libre de
contaminantes en la boquilla para prevenir la pérdida o ganancia de partículas. No tapar el extremo de la
sonda de forma ajustada mientras se enfría el tren de muestreo; puede formarse
vacío en el portafiltro con el resultado no deseado de succionar líquido de los
impactores al filtro.
10.3.1.2 Antes de mover el
tren de muestreo al sitio de limpieza, retirar la sonda del tren de
muestreo y tapar la abertura de salida,
cuidar de no perder ningún condensado que pudiera estar presente. Tapar la
entrada del filtro donde se conecta la sonda.
10.3.1.3 Retirar el cordón
umbilical del último impactor y tapar el impactor. Tapar la salida del
portafiltro y la entrada del impactor.
Utilizar siempre tapones libres de contaminantes como: plástico, vidrio, cera o
cinta de PTFE para cerrar las aberturas.
10.3.1.4 Como alternativa,
el siguiente procedimiento puede ser utilizado para desensamblar el tren antes
que la sonda y el portafiltro se enfríen completamente:
10.3.1.5 Inicialmente
desconectar la unión de portafiltro a impactor, tapar las salidas sin apretar.
Posteriormente desconectar la sonda del portafiltros o ciclón, tapar las
salidas sin apretar. Tapar el extremo de la sonda y retirar el cordón umbilical
como se describe en 10.3.1.3
10.3.1.6 Transferir la
sonda y el ensamble filtro-impactor a un área limpia y protegida del viento y
de otras causas potenciales de contaminación o pérdida de muestra.
10.3.1.7 Inspeccionar el
tren antes y durante el desensamble, anotar cualquier condición anormal. Tener
precaución especial para garantizar que todos los instrumentos necesarios para
recuperar la muestra no provoquen contaminación.
10.4 Recuperación de
las muestras, tratar la muestra de la siguiente forma: ver diagrama de
recuperación de muestras.
10.4.1 Recipiente No.1 (Filtro de la muestra).- Retirar
cuidadosamente el filtro del portafiltro y colocarlo en un caja Petri
previamente identificada. Para manejar el filtro, usar pinzas de polipropileno
lavadas con ácido o recubiertas de PTFE, o guantes de cirujano desechables que
se encuentren limpios, lavados con agua y secos. Si es necesario doblar el
filtro, cuidando que las partículas queden dentro del doblez. Cuidadosamente,
transferir el filtro y cualquier partícula o fibras de filtro adheridas al
soporte del filtro a una caja Petri usando un cepillo de cerdas de nylon
(previamente lavado con ácido y seco). No utilizar materiales metálicos. Sellar
la caja Petri e identificarla.
10.4.2 Recipiente 2. (Lavado de acetona). Realizar este
procedimiento solamente si se determinan partículas. Cuantitativamente
recuperar las partículas y cualquier otro condensado de la boquilla, tubo
interior de la sonda, conexiones y la parte delantera del portafiltros, lavando
estos componentes con un volumen total de acetona de 100 ml, simultáneamente
cuidar que ningún polvo del exterior de la sonda u otra parte se introduzca en
la muestra. Es necesario usar exactamente 100 ml de acetona para la subsecuente
corrección por blanco.
10.4.2.1 Retirar
cuidadosamente la boquilla y limpiar la superficie interna con acetona,
utilizando una pizeta y un cepillo de material no metálico. Cepillar hasta que
el lavado con acetona no presente partículas visibles, hacer un enjuague final
de la superficie interna con acetona.
10.4.2.2 Cepillar y lavar
la muestra presente dentro de las conexiones de la sonda con acetona, de manera
similar, hasta que no presenten partículas visibles. Lavar el tubo interior de
la sonda con acetona por inclinación y rotación mientras se agrega acetona por
la parte superior, de tal forma que toda la superficie interna se humedezca con
acetona. Drenar la acetona por la parte inferior de la sonda hasta el
recipiente de la muestra. Utilizar un embudo para transferir el líquido de la
sonda al recipiente. Continuar el lavado con acetona y con un cepillo no
metálico. Sostener la sonda en una posición inclinada, añadir acetona por la
parte superior de la sonda cepillando con una acción giratoria tres veces a
través de la sonda. Sostener el recipiente para la muestra por la parte
inferior de la sonda y recuperar la acetona y cualquier partícula que se desprenda
a través de la sonda, hasta que ninguna partícula y/o residuos sean observados
en la superficie del tubo interior.
10.4.2.3 Enjuagar el
cepillo con acetona, y cuantitativamente colectar los lavados en el recipiente.
Después del lavado, enjuagar por última vez la sonda como se describe en la
sección anterior.
10.4.2.4 Se recomienda que
dos personas efectúen el procedimiento de limpieza para minimizar la pérdida de
muestra. Entre corridas de muestreo, mantener los cepillos limpios y protegidos
de contaminación. Limpiar el interior de la parte frontal del portafiltro,
frotando la superficie con un cepillo no metálico y lavar con acetona. Lavar
cada superficie tres veces o más si es necesario hasta remover cualquier
partícula visible. Realizar un último lavado del cepillo y el portafiltro.
Después que todos los lavados con acetona y las partículas han sido colectados
en el recipiente para muestra, cerrarlo de tal manera que se evite la pérdida
de acetona durante el transporte al laboratorio.
10.4.2.5 Marcar la altura
del nivel del líquido en frasco que contiene la muestra, con el fin de detectar
pérdidas durante el transporte. Identificar claramente el recipiente de la
muestra.
10.4.3 Recipiente No. 3
(Lavado de la sonda). Mantener la sonda ensamblada limpia y libre de
contaminación durante su lavado. Lavar la boquilla, conexiones, tubo interior y
parte delantera del portafiltro completamente con un total de 100 ml de HNO3 0,1N, colocar la
solución de lavado en un recipiente identificado claramente.
Nota: El uso de 100 ml
exactos es necesario para la corrección de blancos, marcar el volumen del
líquido en cada recipiente para determinar pérdidas durante el transporte.
Sellar el recipiente e identificar claramente el frasco.
10.4.3.1 Por último, lavar
la boquilla, tubo interior y parte delantera del portafiltro con agua seguido
de acetona, descartar estos lavados.
10.4.4
Recipiente No. 4 (Impactores 1 a 3). Medir el líquido en el impactor No. 1, con
una precisión de 0,5 ml usando una probeta. Registrar el volumen. Esta
información es necesaria para calcular el contenido de humedad de los gases de
la fuente fija. Lavar cada uno de los tres impactores, el soporte del filtro y
la parte posterior del portafiltro y las conexiones de vidrio, lavar
completamente con 100 ml de HNO3 0.1N.
10.4.4.1
Debido a que estos impactores pueden presentar grandes volúmenes de líquido, se
pueden almacenar las soluciones de los impactores 1 a 3 en más de un
recipiente, si es necesario.
Nota: El
uso de 100 ml exactos es necesario para la corrección de blancos, marcar la
altura del líquido en cada recipiente para determinar pérdidas durante el
transporte. Sellar el recipiente e identificar claramente el contenido.
10.4.5
Recipiente No. 5ª (HNO3
0,1N), 5B (KMnO4/H2SO4 solución
de captura) y 5C (HCl 8 N, solución de lavado y dilución).
10.4.5.1 Si
en las muestras se analiza mercurio, verter todo el contenido del impactor
(usualmente el No. 4), que
inmediatamente precede a los impactores con permanganato, a una probeta, medir
el volumen con una precisión de 0,5 ml. Esta información es necesaria para
calcular el contenido de humedad del gas.
10.4.5.2
Colocar el líquido en el recipiente 5 A. Lavar el impactor con exactamente 100
ml de HNO3
0,1N, verter el lavado en el recipiente No. 5A.
10.4.5.3
Verter todo el líquido de los impactores con permanganato en una probeta, medir
el volumen con una precisión de 0,5 ml. Esta información es necesaria para
calcular el contenido de humedad del gas de la fuente fija. Colocar esta
solución dentro del recipiente No. 5B. Usando un volumen exacto de 100 ml de
solución acidificada de KMnO4 limpia, para todos los lavados, (aproximadamente 33
ml, por cada lavado), lavar los dos impactores con permanganato y conexiones de
vidrio un mínimo de tres veces. Verter los lavados en el contenedor 5B,
asegurarse de transferir todo el material precipitado de los dos impactores que
se encuentre suspendido. De igual forma, utilizar un total de 100 ml de agua
para lavar los impactores con permanganato y conexiones de vidrio, un mínimo de
tres veces, verter los lavados en el contenedor 5B, asegurarse de transferir
las partículas suspendidas. Marcar la altura del líquido en el recipiente,
sellarlo, identificar claramente el contenido.
10.4.5.4 Si
no hay depósitos remanentes visibles, después del lavado con agua, no es
necesario ningún lavado posterior. Sin embargo, si se presentan depósitos en la
superficie de los impactores, lavarlos con 25 ml de HCl 8N, verter los lavados
en un recipiente para muestras separado, identificarlo con el número 5C,
conteniendo 200 ml de agua. Primero, añadir 200 ml de agua en el recipiente,
posteriormente, lavar las paredes y el tubo interno del impactor con HCl,
girando el impactor por un costado y girándolo de tal manera que el HCl entre
en contacto con la superficie interna. Usar solamente un total de 25 ml de HCl
8,0 N para lavar ambos impactores de permanganato combinados.
10.4.5.5
Lavar el primer impactor, posteriormente verter el lavado en el segundo
impactor para lavarlo. Finalmente, verter los 25 ml de HCl 8N utilizados en el
recipiente. Marcar la altura del líquido en el recipiente para identificar
pérdidas durante el transporte.
10.4.6
Recipiente No. 6 (Sílica gel). Observar el color de la sílica gel para
determinar si ha sido utilizada completamente, escribir la anotación pertinente
en la bitácora. Transferir la sílica gel de su impactor a su recipiente
original y séllelo. Puede usarse un embudo para facilitar esta operación, no
usar agua ni otro tipo de líquido, debido a que la ganancia de peso de la
sílica gel se utiliza para calcular la humedad, como alternativa, utilizar una
balanza granataria para determinar el peso de sílica gel, redondeando al 0.5 g más cercano.
10.4.7
Recipiente No.7 (Blanco de acetona).
Si se determinan partículas, por lo menos una vez, durante el muestreo, verter
100 ml de la acetona utilizada para el proceso de recuperación en el recipiente
No. 7, identificarlo y sellarlo.
10.4.8
Recipiente No. 8 A, (blanco de HNO3 0,1N). Por lo menos, una vez durante el
muestreo, agregar 300 ml de la solución de HNO3 0,1N utilizada en el proceso de recuperación
de la muestra en el recipiente No. 8A,
identificar y sellar el recipiente.
10.4.9
Recipiente 8B (Blanco de agua). Por lo menos, una vez durante el muestreo, colocar
100 ml del agua utilizada en el proceso de recuperación de la muestra en el
recipiente No. 8B, identificar y sellar el recipiente.
10.4.10
Recipiente No. 9 (Blanco de solución 5% de HNO3/10% H2O2). Por lo menos, una vez durante el muestreo,
verter 200 ml de solución 5% de HNO3/10% H2O2 utilizada en el impactor de ácido nítrico de
la muestra en el recipiente No. 9, identificar y sellar el recipiente.
10.4.11
Recipiente No. 10 (Blanco de solución de KMnO4 acidificada). Por lo menos, una vez durante el
muestreo, verter 100 ml de solución de KMnO4 acidificada utilizada como solución en los
impactores y en el proceso de
recuperación de muestra en el recipiente No. 10, identificar y sellar el
recipiente.
10.4.12
Recipiente No. 11 (Blanco de HCl 8,0 N). Por lo menos, una vez durante el
muestreo, verter 200 ml de agua en un recipiente identificado como No. 11,
posteriormente, agregar con agitación 25 ml de HCl 8,0 N, mezclar, identificar
y sellar el recipiente.
10.4.13
Recipiente No. 12 (Blanco de filtro). Por lo menos, una vez durante el
muestreo, colocar en una caja Petri identificada como No.12, tres filtros no usados del mismo lote que los filtros
utilizados en el muestreo. Sellar la caja Petri.
11.
Cálculos
11.1
Cálculos de muestreo.
11.1.1 Masa
total de agua recolectada en tren de impactores (en estado líquido).
Ecuación
No. 10
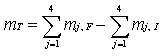
Donde:
mT = Masa
total de agua recolectada en tren de impactores, (g).
mj,I = Masa Inicial del impactor ‘j’, (g)
mj,F = Masa Final del impactor ‘j’, (g)
11.1.2 Condiciones
promedio de presión y temperatura en gasómetro.
Ecuación
No. 11

Donde:
PG,PROM = Presión promedio en gasómetro, Pa
PB = Presión barométrica, Pa
DHi = Caída de presión en medidor de orificio en el punto de muestreo
‘i’, pulgadas H2O
N = Número de
puntos de muestreo, adimensional
Ecuación
No. 12
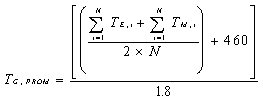
Donde:
TG,PROM = Temperatura promedio en gasómetro, ºK
TE,i = Temperatura de entrada a gasómetro en punto ‘i’, ºF
TM,i = Temperatura de salida de gasómetro en el punto ‘i’, ºF
N = Número
de puntos de muestreo, adimensional
11.1.3 Cálculo del Volumen de Agua Recolectada a
Condiciones de Gasómetro (en estado gaseoso).
Ecuación
No. 13
![]()
Donde:
VH2O,G = Volumen de Agua Recolectada a
Condiciones de Gasómetro, lt
11.1.4. Cálculo del Volumen Real de Gas Seco
Muestreado (cuando las pruebas de hermeticidad fueron aprobadas).
Ecuación
No. 14
![]()
Donde:
?VG,REAL = Volumen real de gas seco muestreado a condiciones de gasómetro,
(lt)
VG,I = Volumen Inicial de Gasómetro, ft3
VG,F = Volumen Final de Gasómetro, ft3
= Coeficiente
de Calibración de gasómetro, adimensional
11.1.5
Cálculo del Contenido de Humedad en el Gas Obtenido en el Muestreo (%HM).
Ecuación No. 15
![]()
Donde:
%HM = Contenido de humedad en el gas obtenido en el muestreo, (%
volumen).
11.1.6
Cálculo del Contenido de Humedad de Saturación (%HS).
Estimar la humedad de saturación a las condiciones
promedio de los gases en el ducto (TC,PROM) y su presión
absoluta promedio (PC,PROM).
11.1.6.1 Cálculo del Contenido de Humedad (%H). El contenido de humedad real en la
muestra es aquel que resulte menor entre HM y HS.
Ecuación No. 16
![]()
Donde:
%H = Contenido de Humedad en
el Gas, [% volumen].
11.1.6.2
Cálculo de la Temperatura Promedio de Gas en el Ducto (TC,PROM).
Ecuación No. 17
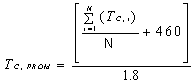
Donde:
TC, PROM = Temperatura Promedio del Gas,
ºK
TC, i = Temperatura
del Gas en Punto Transversal "i", ºF
N = Número Total de Puntos Transversales
11.1.7
Cálculo de la Presión Absoluta Promedio de Gas en el Ducto (PC,PROM).
Para esta sección PC,PROM deberá ser
expresada en Pa.
11.1.8
Cálculo de la Presión Dinámica Promedio (DPPROM).
Ecuación No. 18 y No. 19
![]()
![]()
Donde:
DPPROM = Presión Dinámica Promedio, Pa
?Pi = Presión Dinámica en Punto Transversal "i", “H2O
N = Número Total de Puntos Transversales.
11.1.9 Cálculo del Peso Molecular Base Húmeda (MBH).
Ecuación No. 20
![]()
Donde:
MBH = Peso molecular promedio base húmeda, g/gmol o lb./lbmol
MBS = Peso molecular promedio base seca, g/gmol o lb./lbmol
H = Contenido de Humedad, fracción en volumen
11.1.10
Cálculo de la Velocidad Promedio del Gas (VPROM).
Ecuación No. 21
![]()
Donde:
VPROM = Velocidad
Promedio del Gas, m/s
TC, PROM = Temperatura Absoluta Promedio del Gas, ºK
PC, PROM = Presión Absoluta Promedio del
Gas, Pa
?PPROM = Presión Dinámica Promedio, Pa
CP = Coeficiente de calibración de Tubo de Pitot,
Adimensional
MBH = Peso molecular promedio base húmeda, g/gmol
o lb./lbmol
11.1.11 Cálculo del Isocinetismo Global (%I).
Ecuación No. 22
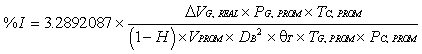
Donde:
%I = Por ciento de Isocinetismo global, adimensional
VPROM = Velocidad promedio del gas, m/s
?VG,REAL = Volumen real de
gas seco muestreado a condiciones de gasómetro, lt
TC, PROM = Temperatura
absoluta promedio del Gas, ºK
PC, PROM = Presión
absoluta promedio del Gas, Pa
TG, PROM = Temperatura
Absoluta Promedio de Gasómetro, ºK
PG, PROM = Presión
Absoluta Promedio de Gasómetro, Pa
qT = Tiempo Total de Muestreo, minutos
DB = Diámetro de boquilla, en pulgadas
MBH = Peso molecular promedio base húmeda, g/gmol o lb./lbmol
Criterios
de Aceptación de Prueba.
|
Criterio de aceptación de prueba de: 90 < %I <
110 |
11.1.12 Cálculo de la Concentración de Partículas
Suspendidas Totales.
Ecuación
No. 23

Donde:
CPST,CNBS = Concentración
de partículas suspendidas totales a condiciones normales Base Seca, mg/m3
mPST,FILTRO = Masa de
partículas recolectada en filtro, mg.
mPST,LAVADO = Masa de
partículas recolectada en lavado, mg.
CPST,BLANCO = Concentración
de partículas en blanco de medio de lavado, mg/ml.
?LAVADO = Volumen de
medio de lavado, ml.
?VG,REAL
= Volumen real de gas seco muestreado a
condiciones de gasómetro, lt
TG,PROM = Temperatura
Promedio de Gasómetro, ºK
PG,PROM = Presión
Promedio de Gasómetro, Pa
11.1.13 Cálculo del Flujo Volumétrico de Gases en
el Ducto.
Ecuación
No. 24
![]()
Donde:
QDUCTO,CNBS = Flujo
volumétrico de gases en el ducto a condiciones normales base seca, m3/min.
VPROM = Velocidad
promedio de gases, m/s
AT = Area transversal interna del ducto, m2
TC,PROM = Temperatura
promedio de gas en ducto, ºK
PC,PROM = Presión
promedio de gas en ducto, Pa
11.1.14 Cálculo del Flujo Másico de Partículas
Suspendidas Totales en el Ducto.
Ecuación No. 25
![]()
Donde:
EPST = Flujo másico de partículas en ducto, Kg./h
QDUCTO,CNBS = Flujo
volumétrico de gases en el ducto a condiciones normales base seca, m3/min
CPST,CNBS = Concentración
de partículas suspendidas totales a condiciones normales base seca, mg/m3
11.2 Cálculo de la
concentración de metales
11.2.1 Fracción Analítica 1A-Parte Frontal-Excepto
Mercurio.
Ecuación
No. 26
![]()
Donde:
mFH,i = Masa total del metal “i” (excepto Hg), recolectada en la parte
frontal del Tren de muestreo, ( g)
CA1,i = Concentración del metal “i” en
la Fracción Analítica 1A, ( g/ml)
FD = Factor de dilución correspondiente a la secuencia de tratamiento de
la fracción 1A
VSOL,1 = Volumen total de la solución de
muestra digerida (Fracción 1), (ml)
NOTA: Si
las Fracciones Analíticas 1A y 2A se combinan, utilizar alícuotas
proporcionales, y posteriormente realice los cambios adecuados en las
ecuaciones correspondientes a estos cálculos.
11.2.2 Fracción Analítica 2A-Parte Trasera-Excepto
Mercurio.
Ecuación
No. 27
![]()
Donde:
mBH,i = Masa total del metal “i” (excepto Hg) recolectada en la parte
trasera del Tren de Muestreo, (g)
CA2,i = Concentración
del metal “i” en la Fracción Analítica 2A, (g/ml)
FA = Factor de dilución correspondiente a la secuencia de
tratamiento de la Fracción 2A
VA = Volumen Total de la Solución de Muestra Digerida (Fracción 2A), (ml)
11.2.3 Masa
Total de Metales en el Tren de Muestreo-Excepto Mercurio
Ecuación No. 28
![]()
Donde:
mi = Masa
total del metal “i” (excepto Hg) en todo el Tren de Muestro, (g)
mFH,i = Masa
total del metal “i” (excepto Hg) recolectada en la parte frontal del Tren de
Muestreo, (g)
mBH,i = Masa
total del metal “i” (excepto Hg) recolectada en la parte trasera del Tren de
Muestreo, (g)
mFH,i,BLANCO = Masa total
del metal “i” (excepto Hg) recolectada en la parte Frontal del Tren de Muestreo
para el Blanco Analítico, (g)
(Ver Ajuste de Blancos para Metales Excepto
Hg)
mBH,i,BLANCO = Masa total
del metal “i” (excepto Hg) recolectada en la parte trasera del Tren de Muestreo
para el Blanco Analítico, (g)
(Ver Ajuste de Blancos para Metales Excepto
Hg)
NOTA:
Ajuste de Blancos para Metales Excepto Hg:
11.2.4 Cálculo en la parte frontal:
Ecuación No. 29
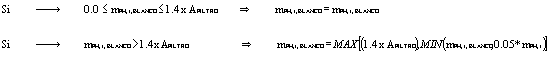
Donde:
Todas
las masas están en g.
AFILTRO = Area
de Filtro, (pulg²)
11.2.5 Cálculo en la parte trasera:
Ecuación No. 30
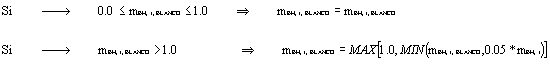
Donde:
Todas
las masas están en g.
11.2.6 Fracción Analítica 1B-Parte Frontal-Mercurio.
Ecuación No. 31
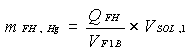
Donde:
mFH,Hg = Masa
total de mercurio recolectada en la parte frontal del Tren de Muestreo, (g)
QFH = Masa
total de mercurio en la alícuota seleccionada para digestión y análisis de la
Fracción Analítica 1B, (g)
VSOL,1 = Volumen total de la
solución de muestra digerida (Fracción 1), (ml)
VF1B = Volumen de
alícuota analizada de la Fracción 1B (ml)
11.2.7 Fracciones Analíticas 2B, 3A, 3B y 3C-Parte
Trasera-Mercurio.
Ecuación No. 33
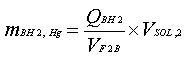
Donde:
mBH2,Hg = Masa total de
mercurio recolectada en la Fracción 2, (g)
QBH2 = Masa total
de mercurio en la alícuota seleccionada para digestión y análisis de la
Fracción Analítica 2B, (g)
VSOL,2 = Volumen total
de la solución de muestra digerida (Fracción 2), (ml)
VF2B = Volumen de
la Fracción 2B, (ml)
Ecuación
No. 34
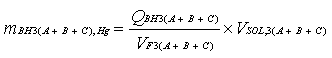
Donde:
mBH3(A+B+C),Hg = Masa total de mercurio recolectada por separado en las Fracciones
3A, 3B
o 3C, (g)
QBH3(A+B+C) = Masa
total de Mercurio en la Alícuota seleccionada para digestión y análisis de las Fracciones Analíticas 3A, 3B o 3C, (g)
VSOL,3(A+B+C) = Volumen
total, separado, de las Fracciones Analíticas 3A, 3B o 3C, (ml)
VF3(A+B+C) = Volumen
analizado, separado, de las Fracciones Analíticas 3A, 3B o 3C, (ml)
Ecuación No. 35
![]()
Donde:
mBH,Hg = Masa total de mercurio recolectada en la
parte trasera del tren de muestreo, (g)
mBH2,Hg = Masa
total de mercurio recolectada en la Fracción 2, (g)
mBH3(A+B+C),Hg = Masa total de
mercurio recolectada por separado en las Fracciones 3A, 3B
o 3C, (g)
11.2.8 Masa Total de Mercurio en el Tren de
Muestreo.
Ecuación No. 36
![]()
Donde:
mHg = Masa total de mercurio en todo el tren de
Muestro, (g)
mFH,Hg = Masa total de mercurio recolectada en la
parte frontal del Tren de Muestreo, (g)
mBH,Hg = Masa total de mercurio recolectada en la
parte trasera del Tren de Muestreo, (g)
mFH,Hg,BLANCO = Masa total de mercurio recolectada en la
parte Frontal del Tren de Muestreo para el Blanco
Analítico, (g) (Ver Ajuste de Blancos para Mercurio)
mBH,Hg,BLANCO = Masa total de mercurio recolectada en la
parte trasera del Tren de Muestreo para el Blanco
Analítico, (g) (Ver Ajuste de Blancos para Mercurio)
NOTA:
Ajuste de Blancos para Mercurio:
Ecuación 37
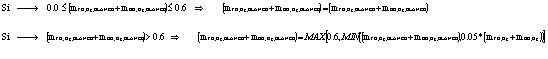
Donde:
Todas
las masas están en g.
11.2.9 Cálculo de Concentración de Metales.
Ecuación 38
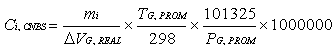
Donde:
Ci,CNBS = Concentración del Metal ‘i’ a Condiciones
Normales Base Seca, mg/m3
mi = Masa del Metal ‘i’ Colectada en el Tren de
Muestreo, g
?VG,REAL = Volumen Real de Gas Seco Muestreado a
Condiciones de Gasómetro, lt
TG,PROM = Temperatura Promedio de Gasómetro, ºK
PG,PROM = Presión Promedio de Gasómetro, Pa
11.2.10 Cálculo del Flujo Másico.
Ecuación
No. 39
![]()
Donde:
Ei = Flujo másico del metal ‘i’ en el ducto, kg/h
QDUCTO,CNBS = Flujo volumétrico de gases en el ducto a
condiciones normales base seca, m3/min
Ci,CNBS = Concentración del metal ‘i’ a condiciones normales base seca, mg/m3
11.2.11 Mínimos de Detección.
Cuando
el resultado del análisis del metal, da por debajo del mínimo detectable del
equipo utilizado (Absorción y/o Plasma), se deberá reportar la concentración y
emisión del metal como ‘menor a’ el resultado del mínimo detectable expresado
como concentración a condiciones de chimenea. Por ejemplo, si el mínimo
detectable del Plomo es de 0.1 g/ml en la curva
de calibración del espectrofotómetro, y el resultado de un definitivo da por
debajo de este valor en ambas Fracciones (1A y 2A), la concentración y emisión
del metal se calcularían considerando las masas de los blancos como cero, de la
siguiente manera:
![]()
![]()
![]()
![]()
![]()
11.2.12 Blancos Menores al Mínimo de Detección.
Cuando
el Blanco de algún metal resulte por debajo del mínimo detectable en alguna
fracción, su masa deberá ser considerada con magnitud de cero en dicha
fracción, para fines de cálculos de concentración y emisión de dicho metal.
Cuando
un metal fue detectado por debajo de su mínimo detectable en alguna fracción, y
el blanco por alguna razón presenta un valor por encima del mínimo detectable,
la masa del blanco deberá ser considerada con magnitud de cero en dicha
fracción, para fines de cálculos de concentración y emisión de dicho metal, y
seguir los cálculos indicados en el inciso 11.2.11.
11.2.13 Criterios para Promedios.
Dado
que generalmente un muestreo de metales se compone de más de una muestra
definitiva, es necesario establecer los criterios a utilizar para el cálculo
del promedio global de concentración y emisión obtenidas en todos los
resultados. Para lo anterior: (a) el promedio de concentración y emisión de
algún metal equivale al promedio aritmético de todos los definitivos
ejecutados; (b) en caso de que uno o más de los definitivos posean un resultado
‘menor a’ (equivalente al mínimo detectable de la curva de absorción, expresado
a condiciones de chimenea), y uno o más de los definitivos posean un resultado
normal, para el promedio global se deberán de considerar los resultados ‘menor
a’ con magnitud de cero, y; (c) en caso de que todos los definitivos posean un
resultado ‘menor a’, se deberá calcular su promedio con la magnitud indicada y
expresar este promedio como ‘menor a’.
12. Seguridad
12.1 No se ha
determinado la carcinogenicidad de todos los reactivos con precisión, por lo
que cada sustancia química debe tratarse como potencialmente peligrosa para la
salud. La exposición a estas sustancias debe reducirse al menor nivel posible.
12.2 Este método puede
no mencionar todas las precauciones de seguridad asociadas con su uso. El
laboratorio es responsable de mantener un ambiente de trabajo seguro y un
archivo de las normas de seguridad respecto a la exposición y manejo seguro de
las sustancias químicas especificadas en este método.
13. Manejo de residuos
13.1 Es
responsabilidad del laboratorio cumplir con todos los reglamentos federales,
estatales y locales referentes al manejo de residuos, particularmente las
reglas de identificación, almacenamiento y disposición de residuos peligrosos.
13.2 Cada laboratorio
debe contemplar dentro de su programa de control de calidad el destino final de
los residuos generados durante el muestreo.
13.3 Todas las
muestras que cumplan con la Norma de descarga al alcantarillado en el mismo
sistema.
14. Bibliografía
14.1 Determination of
metals emissions from stationary sources, “Emission measurement technical
information center EMTIC TM -029, Environmental Protection Agency, April 1996.
15.
Diagramas
15.1 Diagrama de flujo
para recuperación de muestras.

ANEXO 5A
METODO PARA
DETERMINAR POLICLORODIBENZO-p-DIOXINAS (PCDDs) Y POLICLORODIBENZOFURANOS
(PCDFs)
EN EMISIONES DE
FUENTES ESTACIONARIAS POR CROMATOGRAFIA DE GASES DE ALTA RESOLUCION ACOPLADO
A
ESPECTROMETRIA DE MASAS DE ALTA RESOLUCION (HRGC/HRMS)
Introducción
Este
método está basado en su desempeño, lo cual permite que a futuro no quede
obsoleto, ya que podrá actualizarse siempre y cuando se aplique para la misma
matriz y rango de trabajo.
El
principio de este método se basa en la medición de las tetra-, penta-, hexa-,
hepta- y octacloro dibenzo-p-dioxinas y dibenzofuranos presentes en un extracto
orgánico purificado de la muestra, inyectando una alícuota de 2 µL en el
Sistema HRGC/HRMS.
NOTA: Este método está
basado en su desempeño, se permite que el laboratorio omita cualquier paso o
modifique cualquier procedimiento, suponiendo que todos los requerimientos de
desempeño especificados se cumplan. Al laboratorio no se le permite omitir
cualquier punto de control de calidad, ni los parámetros que se especifiquen
como “NO MODIFICABLES”. Los términos “debe”, “puede” y “deberá” son mencionados
a través de los métodos y están destinados a ilustrar la importancia de los
procedimientos para producir datos verificables en los rangos de trabajo del
método. El término “debe” es usado para indicar que los desarrolladores de este
método encontraron ciertos procedimientos esenciales en muestras analizadas
exitosamente; de todas maneras, estos procedimientos pueden ser modificados u
omitidos si el laboratorio puede demostrar fehacientemente que la calidad de
los resultados no resulta afectada. Los requerimientos para establecer la
equivalencia del método se encuentran en la sección 9.
PARAMETROS NO MODIFICABLES
Cromatógrafo de Gases con Columna Capilar de
Alta Resolución (HRGC)
Espectroscopia de Masas de Alta Resolución
(HRMS)
Contenido
1.
Aplicación y Alcances
2.
Resumen
3.
Definiciones
4.
Interferencias
5.
Seguridad
6.
Equipo y Materiales
7.
Reactivos y Patrones
8.
Recolección, Preservación y Almacenamiento de Muestras
9.
Control de Calidad
10.
Calibración
11.
Procedimiento
12.
Cálculos
13.
Desempeño del Método
14.
Prevención de la Contaminación
15.
Manejo de Residuos
16.
Bibliografía
17.
Tablas y Figuras
1.
Aplicación y Alcances
1.1
Aplicación
Este método aplica para la determinación en
emisiones de fuentes estacionarias industriales de policlorodibenzo-p-dioxinas
y policlorodibenzofuranos (PCDDs/PCDFs), está basado en el muestreo isocinético
de las corrientes de gases de las fuentes estacionarias para posterior
determinación analítica por cromatografía de gases alta resolución acoplada a
espectrometría de masas de alta resolución (HRGC/HRMS).
1.2
Alcance
Los analitos determinados por este método se
encuentran listados en la tabla 17.1.,
La sensibilidad de este método depende del
nivel de las interferencias de cada matriz.
Este método está restringido para utilizarse
sólo bajo la supervisión de analistas expertos en el uso de cromatografía de
gases y en la interpretación de sus resultados. Cada analista debe demostrar la
habilidad para generar resultados aceptables con este método usando los
procedimientos establecidos en la Sección 9.
2.
Resumen
2.1 Una
muestra es extraída isocinéticamente de la corriente de gas (emisión), se
recolecta con una sonda en un filtro de fibra de vidrio y sobre una columna
empaquetada de material adsorbente, la muestra no puede separarse en partículas
y en fracciones de vapor, las dioxinas y furanos son extraídas de la muestra,
separadas por cromatografía de gases de alta resolución y medidas por
espectrometría de masas de alta resolución.
2.2 Si
se encuentran interferencias, el método provee procedimientos de limpieza
selecta para auxiliar al analista en su eliminación.
2.3 Una
cantidad específica (Tabla 17.2) de
muestra se adiciona con una solución que contenga cantidades específicas de
cada uno de los nueve marcadores isotópicos (13C12) de PCDDs y PCDFs listados en la columna 1 de
la tabla 17.3. La muestra es
entonces extraída de acuerdo al procedimiento específico para la matriz. Las
muestras sólidas que muestren una fase acuosa, son filtradas, la fase sólida
(incluyendo filtros) y la fase acuosa se extrae separadamente y los extractos
se combinan antes del procedimiento de limpieza. La extracción se lleva a cabo
con tolueno.
2.4 Los
extractos son sometidos a un tratamiento de lavado ácido-base y secados.
Seguido por un paso de intercambio de solvente, los extractos se limpian en una
columna cromatográfica de alúmina, sílica gel y carbón activado.
2.5 La
preparación del extracto final para el análisis HRGC/HRMS se completa por la
adición, al concentrado AX-21/Celite 545® (o equivalente) columna de elución,
10 a 50 µL (dependiendo del tipo de matriz) de una solución de nonano que
contenga 50 pg/µL de cada uno de los dos estándares de recuperación 13C12-1,2,3,4-TCDD y 13C12-1,2,3,7,8,9-HxCDD
(Tabla 17.3). El primero es usado
para determinar el porcentaje de recuperación de los isómeros tetra- y
pentaclorado de PCDDs/PCDFs, mientras que el último se usa para determinar el
porcentaje de recuperación de los isómeros hexa-, hepta- y ocataclorado de
PPCDDS/PPCDFS.
2.6 De
uno a dos µL del extracto concentrado se inyectan en un sistema HRGC/HRMS capaz
de desarrollar un monitoreo de ión selectivo a un poder de resolución de al
menos 10,000 (definición del valle del 10%).
2.7 La
identificación de OCDD y nueve de los quince isómeros 2,3,7,8-sustitutos (Tabla
17.4), para los cuales se dispone un
estándar 13C-marcado
en la muestra fortificada y las soluciones de los estándares de recuperación
(tabla 17.3), se basa en su elución
a un tiempo de retención exacto (dentro de 0,005 unidades de tiempo de
retención medido en la rutina de calibración) y la detección simultánea de los
dos iones más abundantes en la región del ión molecular. Los seis isómeros
remanentes 2,3,7,8-sustituto (por ejemplo, 2,3,4,7,8-PeCDF; 1,2,3,4,7,8-HxCDD;
1,2,3,6,7,8-HxCDF; 1,2,3,7,8,9-HxCDF; 2,3,4,6,7,8-HxCDF y
1,2,3,4,7,8,9-HPPCDFs), para los cuales no hay disponible estándares internos
de carbón marcado para la solución fortificación de la muestra, y todos los
otros isómeros identificados PPCDDS/PPCDFS se identifican por sus tiempos de
retención relativos cayendo dentro de sus intervalos de tiempo de retención
PPCDDS/PPCDFS respectivo, como se estableció a partir de los datos de la
calibración de rutina y de la detección simultánea de los dos iones más
abundantes en la región molecular del ión. La identificación del OCDF se basa
en su tiempo de retención relativa a 13C12-OCDD y de la detección simultánea de los dos
iones más abundantes en la región molecular del ión. La confirmación se basa en
una comparación de las proporciones de la integración de la abundancia del ión
de las especies del ión molecular a sus proporciones de la abundancia teórica.
2.8 La
cuantificación total de los isómeros individuales, PCDDs y PCDFs se determina
en una curva de calibración de cinco niveles para cada homólogo, durante la
cual cada solución de calibración es analizado a la vez.
3.
Definiciones
Las definiciones presentadas en esta sección
son específicas para este método, pero han sido conformadas para que sean en lo
posible de uso común.
3.1
Blanco de Calibración.- Volumen de agua reactivo que se utiliza para calibrar
el instrumento. El blanco de calibración es un estándar cero.
3.2
Blanco de Campo.- Consiste en un filtro de fibra de vidrio, libre de compuestos
orgánicos con un 99,95% de eficiencia (<0,05 de por ciento de penetración),
empacada para el muestreo, y tratada como una muestra en todos los aspectos,
incluyendo el contacto con los equipos de campo y expuesta a las condiciones
del sitio de muestreo, almacenaje, preservación y todos los procedimientos analíticos.
El propósito del blanco de campo es identificar si en el procedimiento de
muestreo o durante el transporte ocurrió alguna contaminación de la muestra.
3.3
Blanco de Reactivos.- Es una matriz libre de analitos a la cual se le agregan
todos los reactivos en los mismos volúmenes o proporciones usados en el
procesamiento de la muestra. El blanco de reactivos debe llevarse a través de
la preparación de la muestra y el procedimiento analítico. El blanco de
reactivos se usa para documentar la contaminación resultante del proceso
analítico.
3.4
Desviación estándar.- Cuando se utiliza este estadístico en el presente método,
se refiere a la desviación estándar de la muestra(s), calculada a partir de n-1
y no a la de la población (ó) la cual se calcula a partir de n.
3.5
Estándar de Calibración.- Solución preparada de un estándar diluido y/o una
solución patrón y utilizada para calibrar la respuesta del instrumento con
respecto a la concentración del analito.
3.6
Estándar de Verificación de la Calibración.- Punto medio del estándar de
calibración que es utilizado para verificar la calibración inicial en el
tiempo.
3.7
Límite de Detección del Método (LDM).- Concentración mínima de una analito que
puede identificarse, medirse y reportarse con una confianza de 99% cuando la
concentración del analito es mayor a cero.
3.8
Límite práctico de cuantificación (LPC).- Concentración mínima del analito que
puede determinarse con un nivel de confianza predeterminado en condiciones
rutinarias de operación. Este límite puede establecerse entre 5 a 10 veces el
LDM.
3.9
Matriz Adicionada (MA) y Matriz Adicionada Duplicada (MAD).- Alícuota de una
muestra ambiental para la cual cantidades conocidas de los analitos del método
son añadidas en el laboratorio. Las MA y MAD son analizadas exactamente como
una muestra. Su propósito es la cuantificación del sesgo y la precisión causada
por la matriz de la muestra. Las concentraciones bases de los analitos en la
matriz de la muestra debe determinarse en una alícuota separada y los valores
medidos en las MA y MAD corregidas con las concentraciones base.
3.10
Muestra de Control de Calidad (MCC).- Muestra sintética que contiene todos o un
subgrupo de los analitos del método a una concentración conocida. La MCC se
obtiene de una fuente externa al laboratorio o es preparada de una fuente
diferente de los estándares de la fuente de los estándares de calibración. Se
usa para revisar el desempeño del laboratorio con materiales de prueba
preparados externamente a los procesos normales de preparación.
3.11
Rango de Trabajo.- Rango de la concentración sobre el cual la respuesta del
instrumento para el analito es proporcional.
4.
Interferencias
4.1 Los
solventes, reactivos, materiales de vidrio y otros insumos utilizados durante
el procesamiento de las muestras pueden producir artificios y/o líneas de bases
elevadas causando un error en la interpretación de los cromatogramas. Puede
requerirse la selección específica de reactivos y la purificación de los
solventes por destilación en todo el sistema de vidrio. Cuando sea posible, los
reactivos se limpian por extracción o enjuague de solventes.
4.2 El
uso de reactivos de alta pureza y solventes ayuda a disminuir los problemas de
interferencia. Puede requerirse la purificación de solventes por destilación en
todos los sistemas de vidrio.
4.3 Las
interferencias co-extraídas de la muestra variará considerablemente de fuente a
fuente, dependiendo de los procesos industriales que serán muestreados. Los
PCDDs y PCDFs están algunas veces asociados con otros compuestos clorados
interferentes tales como BPC’s y éteres bifenilos policlorados (PCDPE’s),
naftalenos policlorados y alquildibenzofuranos policlorados los cuales pueden
encontrarse en concentraciones altas de varios órdenes de magnitud que aquellos
analitos de interés. Los tiempos de retención de los analitos marcados deben
verificarse usando estándares de referencia. Estos valores deben corresponder a
los intervalos del tiempo de retención establecidos. Mientras que ciertas
técnicas de limpieza se proporcionan como parte de este método, algunas
muestras pueden requerir de pasos de limpieza adicional para alcanzar límites
de detección menores.
4.4 Las
columnas capilares de alta resolución son usadas para resolver muchos de los
isómeros de PPCDDS y PPCDFS tantos como sean posibles; sin embargo, no se
conoce una columna que resuelva todos los isómeros.
5.
Seguridad
5.1 La
carcinogenicidad de todos los reactivos no ha sido determinada con precisión;
de todas maneras; cada sustancia química debe tratarse como potencial peligro a
la salud. La exposición a estas sustancias químicas debe reducirse al menor
nivel posible. Se sugiere que el laboratorio realice monitoreos de higiene
ocupacional de cada reactivo a los que pueda estar expuesto el analista y que dichos
resultados estén disponibles para los analistas.
5.1.1 Se
ha encontrado que el isómero 2,3,7,8-TCDD es un acnegénico, carcinogénico y
teratogénico en estudios con animales de laboratorio. Este es soluble en agua a
aproximadamente 200 ppt y en solventes orgánicos a 0.14%. Con base en la disponibilidad toxicológica y las propiedades
físicas del 2,3,7,8-TCDD, todos los PCDDs/PCDFs deben manejarse sólo por
personal altamente entrenado cabalmente familiarizado con el manejo y
procedimientos de precaución y los riesgos asociados.
5.1.2 Se
recomienda que el laboratorio maneje soluciones estándares diluidas de los
analitos de este método. Sin embargo, si se preparan soluciones primarias,
deben prepararse en una campana y un respirador de gases tóxicos debe usarse
cuando se manejan altas concentraciones.
5.2 Este
método puede no mencionar todas las precauciones de seguridad asociadas con su
uso. El laboratorio es responsable de
mantener un ambiente de trabajo seguro y un archivo de las normas de seguridad
respecto a la exposición y manejo seguro de las sustancias químicas
especificadas en este método. Debe tenerse un archivo de referencias de las
hojas de información de seguridad el cual debe estar disponible a todo el
personal involucrado en estos análisis.
5.3 Los
PCDDs/PCDFs y muestras que se sospecha contienen estos compuestos deben
manejarse usando esencialmente las mismas técnicas empleadas en el manejo de
material radioactivo o infeccioso. Se requiere de un laboratorio bien ventilado
y de acceso controlado. Cada laboratorio debe desarrollar un programa estricto
de seguridad para el manejo de estos compuestos.
5.3.1
Instalaciones.- Cuando se manejan muestras de tamaño de partícula muy pequeño
(suelos, químicos secos, polvos), todas las operaciones (incluyendo la remoción
de muestras desde los contenedores de las mismas, pesado, transferencia y
mezcla) deben desarrollarse en una campana de extracción en la cual se
demuestre que tiene un adecuado flujo de aire. No se permiten grandes pérdidas
en el sistema de ventilación del laboratorio. No deben inhalarse las soluciones
diluidas utilizadas normalmente en el trabajo analítico.
5.3.2
Equipo de protección.- Deben usarse guantes de plástico ajustables, bata de
laboratorio o delantal, lentes de seguridad o máscara, una campana de
extracción adecuado para trabajo radiactivo. Durante las operaciones analíticas
que pueden producir aerosoles ascendentes o polvos, el personal debe usar
equipos de respiración con filtros de carbón activado. El equipo de protección de
los ojos (preferiblemente cubriendo la cara completa) debe usarse mientras se
está trabajando con muestras expuestas o estándares analíticos puros. Cuando se
sospecha o se conoce que las muestras contienen altas concentraciones de
PCDDs/PCDFs, un juego adicional de guantes puede utilizarse debajo de los
guantes de látex.
5.3.3
Entrenamiento.- Los analistas y todo el personal que custodie las muestras
deberá recibir entrenamiento especial para el manejo de muestras y sustancias
altamente peligrosas.
5.3.4
Higiene personal.- Las manos y los antebrazos deben lavarse completamente
después de cada manipulación y antes de cada alimento.
5.3.5
Confinamiento.- Todos los materiales que hayan tenido contacto con las muestras
que hayan presentado cualquier concentración de dioxinas y furanos deberá
tratarse como residuo peligroso.
5.3.6
Emisiones de Vapores.- Las emisiones de vapores de muestras adicionadas desde
el cromatógrafo de gases (GC) y desde las bombas de agitación en el
espectrómetro de masas (MS) deben pasar a través de una columna de carbón
activado o ser burbujeado a través de una trampa que contenga aceite o
alcoholes de alta ebullición para condensar los vapores de CDD/CDF.
5.3.7
Manejo de residuos.- Todo el personal que tenga contacto con los residuos
producidos del análisis, deberá ser entrenado en el manejo de residuos
peligrosos.
5.3.8
Descontaminación
5.3.8.1
Descontaminación del personal.- Después del trabajo analítico el personal
deberá lavarse manos y antebrazos con un jabón suave y abundante agua
corriente.
5.3.8.2 Materiales de
vidrio, herramientas y superficies.- El cloroetano es el solvente menos tóxico
que ha mostrado ser el más efectivo. Una limpieza satisfactoria puede
completarse con un enjuague con cloroetano, después lavar con cualquier
detergente y agua. Si el material de vidrio se enjuaga primero con solvente,
entonces el lavado con agua puede disponerse en el desagüe. Dado el alto costo
de la disposición, es prudente la disminución de residuos de solventes.
5.3.9 Lavandería.- La
ropa que se conoce está contaminada debe colectarse en bolsas de plástico. Las personas que conduzcan las bolsas y
laven la ropa deben ser avisadas del peligro y entrenadas en el manejo
apropiado. La ropa debe lavarse en una lavadora sin contacto, la lavadora debe
trabajar un ciclo antes de ser utilizada nuevamente.
5.3.10 Prueba de
limpieza.- Un método exitoso para determinar la limpieza de las superficies de
trabajo y los equipos es, limpiar las superficies con un trozo de papel filtro.
La extracción y análisis por GC con un detector de captura de electrones (ECD)
pueden permitir un límite de detección de 0,1 g; el análisis usando este método
puede permitir un límite de detección aún más bajo. Concentraciones menores de
0,1 µg por limpieza indican una técnica de limpieza adecuada; cualquier otra
concentración más alta garantiza una limpieza mayor. Una concentración mayor de
10 µg en la limpieza constituye un potencial de peligro agudo y requiere una
limpieza expedita, además de los equipos o espacios de trabajo, e indica que
han sido empleadas prácticas de trabajo no aceptables.
5.3.11 Equipos de
agitación.- El uso de equipos de agitación para la extracción de muestras
presenta la posibilidad de ruptura de las botellas de extracción y el derrame
de ácidos y solventes orgánicos inflamables. Se sugiere un sistema de
contenedor secundario alrededor del agitador para prevenir la dispersión del
ácido y solventes aun en el caso de una ruptura. La velocidad e intensidad de
la acción de agitación debe también ajustarse para disminuir la posibilidad de
ruptura.
6. Equipo y Materiales
La
mención de marcas, modelos y proveedores de equipos y materiales en este método
se citan debido a que fueron los utilizados para desarrollarlo y solamente
tienen propósitos ilustrativos. Su mención no implica ninguna aprobación
oficial. Puede obtenerse un desempeño equivalente usando otros equipos y
materiales que no hayan sido especificados en este método, pero la demostración
del desempeño equivalente de otros equipos y materiales es responsabilidad del
laboratorio que utilice este método.
Sólo
se mencionan los equipos y materiales que son relevantes en este método
analítico.
6.1 Sistema
Cromatografía de Gases/Espectrometría de Masas.
6.1.1 Cromatógrafo de
Gases.- Cromatógrafo de gases con inyector capilar y detector de captura de
electrones (ECD) con sistema computarizado de registro y procesamiento de
datos.
6.1.2 Espectrómetro de
Masas.- Se especifica un instrumento de alta resolución, utilizando una energía
de electrón de 70 volts (nominal) en el modo de electrón de impacto de
ionización. El sistema debe ser capaz
de monitorear una selección de iones (SIM) para al menos 11 iones
simultáneamente, con un ciclo de 1 seg.
o menos. El tiempo de integración mínimo por SIM es de 50 ms por m/z.
6.1.3 Interfase GC/MS.- Puede usarse
cualquier interfase GC/MS que proporcione una respuesta de calibración
aceptable para cada analito de interés a la concentración requerida y alcance
los criterios de desempeño.
6.1.4
Sistema de datos.- Un sistema de cómputo debe estar en la interfase al
espectrómetro de masas. El sistema debe
permitir la adquisición y almacenamiento continuo de todos los datos obtenidos
a través de la duración del programa cromatográfico.
6.1.5
Equipo de evaporación bajo flujo de nitrógeno.- Equipado con baño de agua
controlado en el intervalo de 30 a 60ºC, instalado en una campana de
extracción.
6.1.6
Balanza capaz de pesar con una exactitud entre 0,01 g y 0,0001 g.
6.1.7
Centrífuga.
6.1.8 Baño
de agua, equipado con cobertura de anillos concéntricos y capaz de controlar la
temperatura dentro ± 2ºC.
6.1.9
Horno de secado.
6.2
Columnas GC.
6.2.1 Con
el propósito de tener una determinación en isómeros específicos para
2,3,7,8-TCDD y para permitir la detección de OCDD/OCDF dentro de un intervalo
de tiempo razonable en un análisis HRGC/HRMS, se recomienda el uso de una
columna capilar de sílica fundida DB-5 de 60 m.
6.2.2
Columnas cromatográficas de vidrio, de 300 mm * 10,5, con aditamento de llave
de PTFE
6.3
Materiales
6.3.1
Pipetas Pasteur.- Desechables de 150-mm de longitud x 5-mm de diámetro interior
6.3.2
Pipetas serológicas desechables de 10 ml para la preparación de la columna de
carbón.
6.3.3
Viales de reactivo de 2 ml, de vidrio ámbar. Estos deben silanizarse antes de
usarse
6.3.4
Embudos de separación de 125 ml y 2 L
6.3.5
Concentrador Kuderna-Danish (K-D).
6.3.6 Tubo
concentrador de 10 ml, graduado con calibración verificada.
6.3.7
Pinzas de vidrio de fondo (con junta de tamaño 19/22) se utiliza para prevenir
la evaporación de los extractos.
6.3.8
Matraz de evaporación de 500 ml, anexo al tubo concentrador con resorte
6.3.9
Columna Snyder tres macro bolas
6.3.10
Perlas de ebullición de PTFE o carburo de silicón de malla aproximada de 10/40,
lavadas con hexano antes de su uso.
NOTA: Las
perlas de ebullición de PTFE, pueden flotar en cloruro de metileno, no pueden
trabajar en presencia de cualquier fase acuosa y pueden impregnarse de
compuestos orgánicos no polares.
6.3.11
Viales de vidrio, 1 dracma (1/8 de onza) (o equivalente métrico).
NOTA: El
reuso del material de vidrio debe minimizarse para evitar un aumento de
contaminación. Todo el material de vidrio que es rehusado debe limpiarse
escrupulosamente tan pronto como sea posible después de su uso, de acuerdo al
siguiente procedimiento: Enjuague el material de vidrio con el último solvente
utilizado en él. Lave con agua y detergente caliente, después enjuague con una
cantidad copiosa de agua corriente y varias porciones de agua grado reactivo. Enjuague
con acetona de alta pureza y hexano y guarde el material de forma invertida o
tapando con papel aluminio enjuagado con solvente en un ambiente limpio.
6.3.12
Papel filtro No. 54 o equivalente.- Se recomiendan filtros de fibra de vidrio o
tapones de lana de vidrio.
6.3.13
Reservorio de solventes de 125 ml diámetro de 12,5 cm, compatible con la
columna de carbón.
6.3.14
Contenedores suficientemente grandes de vidrio o acero inoxidable para mantener
los contenidos de un octavo del contenedor de la muestra.
6.3.15
Guantes ajustables desechables.
6.3.16
Espátulas de acero inoxidable.
6.3.17
Adaptador para tubos concentradores.
6.3.18
Trampa Dean-Stark, 5 o 10 ml, con juntas-T, condensador y matraz de 125 ml.
6.3.19
Extractor líquido-líquido continuo.
6.3.20
Material de vidrio del aparato Soxhlet, matraz 500 ml.
6.3.21
Extracto Sohxlet/Dean Stark (opcional), todo de vidrio, matraz de 500 ml.
6.3.22
Embudo de vidrio capacidad para mantener 170 ml de líquido.
6.3.23
Desecador.
6.3.24
Reservorio de solvente (125 ml), 12,35 cm diámetro, compatible con una columna
de carbón gravitatoria.
6.3.25
Evaporador rotatorio con baño de agua y temperatura controlada.
6.3.26 Lana
de vidrio, extraída con cloruro de metileno, secada y almacenada en un vaso de
vidrio limpio.
6.3.27 Vaso
de extracción de vidrio de 250 ml, con tapa de PTFE.
6.3.28
Matraces volumétricos, clase A de 10 ml a 1,000 ml.
7.
Reactivos y Patrones
Los reactivos que requiere el método deben ser
tipo ACS grado reactivo o plaguicida, a menos que otra cosa se indique.
7.1
Agua.- A menos que se indique otro grado, el agua de referencia debe ser grado
reactivo tipo I ASTM.
7.2
Reactivos de la Columna Cromatográfica.
7.2.1
Alúmina neutral, malla 80/200.
Almacenar en un contenedor cerrado a temperatura ambiente, dentro de un
desecador
7.2.2
Alúmina, ácida AG4, extraída en Soxhlet con cloruro de metileno por 24 horas si
los blancos muestran contaminación y activada por calentamiento en un
contenedor de vidrio cubierto por 24 horas a 190ºC. Conservar en frasco de
vidrio con tapa cubierta con PTFE.
7.2.3
Sílica gel grado alta pureza, tipo 60, malla 70-230; extraída en Soxhlet con
cloruro de metileno por 24 horas si los blancos muestran contaminación, activar
por calentamiento en un contenedor de vidrio cerrado por 24 horas a 190ºC.
Conservar en frasco de vidrio con tapa cubierta con PTFE.
7.2.4
Sílica gel impregnada con hidróxido de sodio. Adicionar una parte (por peso) de
solución NaOH 1M a dos partes (por peso) de sílica gel (extraída y activada),
mezclar con un agitador de vidrio hasta que esté libre de brumos. Conservar en
frasco de vidrio con tapa cubierta con PTFE.
7.2.5
Sílica gel impregnada con ácido sulfúrico (por peso) al 40%. Adicionar dos
partes (por peso) de ácido sulfúrico concentrado a tres partes (por peso) de
sílica gel (extraída y activada), mezclar con un agitador de vidrio hasta que
esté libre de brumos almacenar en frasco de vidrio con tapa de rosca y
contratapa de PTFE.
7.2.6
Celite 545® (Supelco) o equivalente.
7.2.7
Carbón activado grado plaguicida, lavado previamente con metanol secar en vacío
a 110ºC. Almacenar en frasco de vidrio con tapa de rosca y contratapa de PTFE.
7.3
Reactivos.
7.3.1
Acido sulfúrico H2SO4, concentrado,
gravedad específica 1,84.
7.3.2
Hidróxido de sodio NaOH al 20% (p/v) en agua.
7.3.3
Cloruro de sodio NaCl reactivo analítico, 5% (p/v) en agua.
7.3.4
Carbonato de potasio K2CO3 anhidro reactivo
analítico.
7.4
Agente desecador.
7.4.1
Sulfato de sodio (anhidro) Na2SO4.- Purificado por
calentamiento a 400ºC durante 4 horas en un contenedor poco profundo, o por
limpieza previa del sulfato de sodio con cloruro de metileno. Si el sulfato de
sodio se limpia previamente con cloruro de metileno, un blanco de reactivos
debe analizarse, demostrando que éste no interfiere del sulfato de sodio.
7.5
Solventes.
7.5.1
Cloruro de metileno CH12Cl2, alta pureza.
7.5.2
Hexano C6H14. Alta
pureza.
7.5.3
Metanol CH3OH.
Alta pureza.
7.5.4
Nonano C9H20. Alta
pureza.
7.5.5
Tolueno C6H5CH3. Alta pureza.
7.5.6
Ciclohexano, C6H12. Alta
pureza.
7.5.7
Acetona CH3COCH3. Alta pureza.
7.6
Soluciones de calibración de concentración de alta resolución (Tabla 17.5).- Para calibrar el instrumento
preparar cinco soluciones de nonano conteniendo carbones no marcados (totalizando
17) y marcados (totalizando 11) de PCDDs y PCDFs a concentraciones conocidas.
Los intervalos de concentración son homólogos dependientes, con el valor menor
para la dioxina tetraclorada y furano (1,0 pg/µL) y los valores más altos para
los isómeros octaclorados (1,000 pg/µL).
7.6.1
Dependiendo de la disponibilidad de los materiales, estas soluciones de
calibración de concentración de alta resolución pueden obtenerse de USEPA. Sin
embargo, estándares secundarios adicionales deben obtenerse de una fuente comercial
y las soluciones deben prepararse en el laboratorio. La trazabilidad de los
estándares debe verificarse contra las soluciones estándares certificadas. Es
la responsabilidad del laboratorio que las soluciones de calibración adquiridas
o preparadas sean de una concentración adecuada antes de ser utilizadas en el
análisis de muestras.
7.6.2
Almacenar las soluciones de calibración en un minivial de 1-ml a temperatura
ambiente protegidos de la luz.
7.7
Solución de verificación del desempeño de la columna.- Esta solución contiene
los isómeros de la primera y la última elución para cada serie de homólogos de
los isómeros desde el tetra- hasta el heptaclorado. La solución también
contiene una serie de otros isómeros TCDD para el propósito de documentar la
resolución cromatográfica. El 13C12-2,3,7,8-TCDD también está presente. Se requiere que
el laboratorio utilice el nonano como el solvente y ajustar el volumen tal que
la concentración final no exceda 100 pg/µL por congénere. La tabla 17.7 resume la composición cualitativa
(requerimiento mínimo) de esta solución de evaluación del desempeño.
7.8
Solución de fortificación de muestra.- Esta solución con nonano contiene los
nueve estándares internos a las concentraciones nominales que están listados en
la tabla 17.2. La solución contiene
al menos un carbón marcado estándar para cada una de las series homólogas y
éste es usado para medir las concentraciones de las sustancias nativas (note
que el 13C12-OCDD no está
presente en la solución).
7.9
Solución estándar de recuperación.- Esta solución de nonano contiene dos
estándares de recuperación, el 13C12-1,2,3,4-TCDD y 13C12-1,2,3,7,8,9-HxCDD, a una concentración
nominal, de 50 pg/µL por compuesto. De 10 a 50 µL de esta solución será
adicionada a cada extracto de muestra antes del paso de concentración final y
el análisis en el HRGC/HRMS.
7.10
Solución de fortificación para una matriz adicionada.- La solución usada para
preparar las muestras MS y MSD, contiene todos los analitos no marcados
listados en la tabla 17.5
correspondiendo al HRCC3.
8.
Recolección, Preservación y Almacenamiento de Muestras
8.1 Las
muestras deben colectarse de acuerdo al procedimiento de muestreo de emisiones
en fuentes estacionarias anexo al método para determinar dioxinas y furanos por
cromatografía de gases por baja resolución.
8.2 No
se adicione ningún preservador a las muestras
8.3
Mantener las muestras a 4ºC desde el momento de su colecta hasta el momento de
la extracción.
8.4
Todas las muestras deben ser extraídas dentro de los 30 días después de
recolectadas y ser completamente analizadas dentro de los 45 días después de la
colecta.
9.
Control de Calidad
9.1
Aspectos Generales:
Cada laboratorio que utilice este método está
obligado a operar un programa de control de calidad (CC) formal. Los
requerimientos mínimos de este programa consisten en una demostración inicial
de la capacidad del laboratorio para cumplir con las especificaciones de
desempeño del método, además realizar análisis continuos de muestras de control
de calidad (MCC) para demostrar la precisión y exactitud continuas y el
análisis de blancos. El desempeño del laboratorio debe compararse con los
criterios aquí establecidos, con objeto de determinar si los resultados de los
análisis cumplen con las especificaciones de desempeño del método. El analista
debe hacer una demostración inicial de su habilidad para generar una exactitud
y precisión aceptables por este método. Esta habilidad debe realizarse como se
menciona en la sección 9.2.
9.1.1 El
analista debe hacer una demostración inicial de su habilidad para generar una
precisión y exactitud aceptable con este método. Esta habilidad se establece
como se describe en la sección 9.2.
9.1.2 En
reconocimiento de los avances que están ocurriendo en la tecnología analítica y
para permitir al analista superar las interferencias de la matriz de la
muestra, se le permite al analista ciertas opciones, improvisar separaciones o
costos menores de medición. Estas opciones incluyen extracciones,
concentraciones, procedimientos de limpieza alternativos y cambios en columnas
y detectores. Técnicas determinativas alternativas, tales como la sustitución
de espectroscopia o técnicas de inmuno-ensayo y cambios que degraden el
desempeño del método, no se permiten. Si una técnica analítica diferente a las
técnicas especificadas en este método será utilizada, esta técnica debe tener
una especificidad igual o mejor que la especificidad de las técnicas en este
método para los analitos de interés.
9.1.2.1 Cada
vez que se realice una modificación al método o que se cambie el analista
responsable de llevar a cabo esta determinación, el analista designado debe
repetir el procedimiento mencionado en la sección 9.2, si el cambio va a afectar el límite de detección del método
(LDM), el laboratorio debe demostrar que el nuevo LDM es menor que un tercio
del nivel regulatorio o un tercio del LDM de este método, cualquier otro es muy
alto. Si la calibración será afectada por el cambio, el analista debe
recalibrar el instrumento como se indica en la sección 10.
9.1.2.2 Es
obligatorio para el laboratorio mantener los registros de las modificaciones
hechas a este método. Estos registros deben de incluir lo siguiente:
- Los nombres, títulos, direcciones y número
de teléfono de los analistas que ejecutaron los análisis y modificaciones y el
encargado de control de calidad que presenció y verificó los análisis y sus
modificaciones.
- Una lista de los parámetros medidos, por
nombre y número CAS.
- Escrito donde se expliquen las razones para
las modificaciones.
- Los resultados de todas las pruebas de CC
del método modificado comparadas con el método original, incluyendo:
a) Calibración.
b) Verificación de la Calibración.
c) Precisión y Recuperación Inicial.
d) Recuperación de los Compuestos Marcados.
e) Análisis de Blancos.
f) Exactitud establecida.
- La información escrita en las bitácoras
tanto del equipo como del analista, deben incluir los siguientes datos:
g) Identificación de la muestra.
h) Número del lote analítico en el cual se
analizó la muestra.
i) Fecha del análisis.
j) Procedimiento cronológico utilizado.
k) Cantidad de muestra utilizada.
l) Número de muestras de control de calidad
analizadas en el lote.
m) Trazabilidad de las calibraciones de los
instrumentos de medición.
n) Registros de bitácoras, en cintas de
respaldo o en otros respaldos de información.
o) Información cruda reportada por los equipos
o por los analistas.
p) Evidencia de la aceptación o rechazo de los
resultados del lote analítico.
9.1.3 Se
requiere el análisis de blancos para demostrar que no existe contaminación. Los
procedimientos y criterios para el análisis de blancos se describen en las
secciones 9.5 y 11.
9.1.4 El
laboratorio debe adicionar todas las muestras con compuestos marcados para
monitorear el desempeño del método. Esta prueba se describe en la sección 9.3. Cuando los resultados de estas
adiciones indican un desempeño del método atípico para las muestras, las
muestras se diluyen para alcanzar el desarrollo del método dentro de los
límites aceptables.
9.1.5 El
laboratorio debe en una base continua, demostrar a través de la verificación de
la calibración y el análisis de alícuotas de precisión y recuperación continua
que el sistema analítico está en control. Estos procedimientos están descritos
en la sección 11.
9.2 Criterios
del Desempeño del Sistema.- El laboratorio puede usar la columna GC recomendada
en la sección 6.2. Deben
documentarse todos los criterios de desempeño aplicables (especificados en las
Secciones 9.2.1 y 9.2.2) conociéndose antes del análisis
de cualquier muestra desarrollada. La sección 11.4 provee las condiciones del GC recomendadas que pueden ser
usadas para satisfacer los criterios requeridos. La verificación de la columna
GC sólo se requiere al inicio de un periodo de 12 horas durante el cual las
muestras serán analizadas. Un blanco de reactivos corrido en el HRGC/HRMS se
requiere entre la corrida de calibración y la primera muestra corrida. El mismo
blanco de reactivos extraído puede entonces analizarse más de una vez si el
número de las muestras dentro de cada lote requiere más de 12 horas de
análisis.
9.2.1
Desempeño de la Columna GC.
9.2.1.1
Inyectar 2 µL (sección 6.1.1) de la
solución de verificación del desempeño de la columna (sección 7.7) y obtener los datos del monitoreo
de ión selectivo (SIM) como se describe en la sección 11.4.2 dentro de un ciclo de tiempo total de < 1 segundo (sección
11.5.4.1).
9.2.1.2 La
separación cromatográfica entre el 2,3,7,8-TCDD y los picos representando
cualquiera de los otros isómeros TCDD no marcados deben resolverse con un valle
de < = 25% donde:
Ecuación 1:
Porcentaje del valle = (x/y) (100)
donde:
x = medida del 2,3,7,8-TCDD más cercano al
isómero de elución.
y = altura del pico del 2,3,7,8-TCDD.
Es la responsabilidad del laboratorio
verificar las condiciones adecuadas para la resolución apropiada del
2,3,7,8-TCDD de todos los otros isómeros TCDD. La solución de verificación del
desempeño también contiene los primeros y los últimos PCDDs/PCDFs eluados
conocidos bajo las condiciones especificadas en este protocolo. Sus tiempos de
retención son usados para determinar los intervalos del tiempo de
retención de los ocho homólogos que se
usan para propósitos cualitativos y cuantitativos. Todos los picos (que incluye
13C12-2,3,7,8-TCDD)
deben marcarse e identificarse en los cromatogramas. Además, todos los primeros
eluados de una serie de homólogos deben marcarse con la letra F, y todos los
últimos eluados de una serie de homólogos deben marcarse con la letra L.
Cualquier ión seleccionado individual de perfil común (SICP) (para el tetras,
éste será el SICP para el m/z 322 y m/z 304) o el homólogo reconstruido del ión
común (para el tetras, éste corresponderá a m/z 320 + m/z 322 + m/z 304 + m/z
306) constituye una forma aceptable de la presentación de datos. También se
requiere un SICP para los compuestos marcados (por ejemplo, m/z 334 para el
TCDD marcado).
9.2.1.3 Los
tiempos de retención para la transferencia de los iones característicos de una
serie de homólogos a la serie siguiente más alta debe indicarse en el SICP. Es
absolutamente necesario una transferencia exacta en tiempo para monitoreo
exacto de estos compuestos. Una tolerancia permitida en la verificación diaria
con la solución de verificación del desempeño del GC debe ser mejor que 10
segundos para los tiempos de retención absolutos para todos los componentes de
la mezcla. Debe ejercerse especial precaución para el tiempo de transferencia
entre el último congénere tetraclorado (por ejemplo, 1,2,8,9-TCDD) y el primer
congénere pentaclorado (por ejemplo, 1,3,4,6,8-PeCDF), tal que estos dos
compuestos eludan dentro de 15 segundos de cada uno de los otros en la columna
DB-5 60 m. Un laboratorio con un sistema GC/MS que no es capaz de detectar
ambos isómeros (1,2,8,9-TCDD y 1,3,4,6,8-PeCDF) dentro de un análisis debe
tomar acciones correctivas. Si no se usa la columna recomendada, entonces el
primero y el último isómero eluado de cada homólogo debe determinarse
experimentalmente en la columna que es usada, y los isómeros apropiados deben
entonces usarse para la definición del intervalo y los tiempos de
transferencia.
9.2.2
Desempeño del espectrómetro de masas.
9.2.2.1 El
espectrómetro de masas debe operarse en el modo ionización del electrón. Un
poder de resolución estática de al menos 10,000 (10% de la definición del
valle) debe demostrarse a una masa apropiada antes de que cualquier análisis se
lleve a cabo (sección 11.4.2). La
verificación del poder de resolución estático debe desarrollarse al inicio y al
final de cada periodo de 12 horas. Sin embargo, se recomienda que una
verificación de la resolución estática se haga y documente antes y después de
cada análisis. Deben implementarse acciones correctivas en caso de que el poder
de resolución no cumpla con los requerimientos.
9.2.2.2 Los
tiempos cromatográficos para PCDDs y PCDFs exceden la longitud de los términos
de la estabilidad de la masa del espectrómetro de masas. Debido a que el
instrumento se opera en el modo de alta resolución, la masa se deriva de unas
pocas ppm (por ejemplo, 5 ppm en masa) puede tener efectos adversos serios en
el desempeño del instrumento. Por lo tanto, es obligatoria una corrección de
masa derivada. Para este efecto, se recomienda seleccionar un ión de masa más
exacto de los compuestos de referencia (se recomienda PFK) usando para la
afinación del espectrómetro de masas. La selección del ión más exacto es
dependiente de las masas de los iones monitoreados dentro de cada descriptor.
La Tabla 17.6 ofrece algunos iones
sugeridos para obtener la masa más exacta. Sin embargo, un ión más exacto
aceptable a cualquier masa entre el ión más ligero y el más pesado en cada
descriptor puede usarse para monitorear y corregir las masas derivadas. El
nivel del compuesto de referencia (PKF) medido dentro de la cámara de iones
durante los análisis HRGC/HRMS debe ajustarse tal que la amplitud de la mayoría
de las señales de los iones más exactos seleccionados (prescindiendo de los
números descriptores) no excedan el 10% de la escala completa de deflexión para
un grupo dado de parámetros detectados. Bajo esas condiciones, los cambios en
la sensibilidad que pueden ocurrir durante el análisis son efectivamente
monitoreados.
NOTA:
Excesivo PFK (o cualquier otra sustancia de referencia) puede causar problemas
de ruido y contaminación de la fuente de iones resultando en un incremento en
poco tiempo para fuentes de limpieza.
9.2.2.3 La documentación
del poder de resolución del instrumento debe entonces completarse por el
registro de los perfiles de los picos de la señal de referencia masa-alta (m/z
380,9760) obtenidos durante el experimento de marcado de picos de arriba por el
uso de la masa baja del ión PFK a una referencia de m/z 304,9824. El poder mínimo de resolución de
10,000 debe demostrarse en la masa-alta del ión mientras se transmite a un voltaje
de aceleración más bajo que el ión de referencia masa-baja, el cual es
transmitido a una sensibilidad completa. El formato del perfil del pico
representado debe permitir la determinación manual de la resolución, por
ejemplo, el eje horizontal debe estar a una escala de masa equilibrada (uma o
ppm por división). El resultado del pico ancho medido (desarrollado a 5% del
máximo, el cual corresponde al 10% de la definición del valle) debe aparecer en
la copia dura y no puede exceder 100 ppm a m/z 380,9760 (o 0,038 uma a esa masa
particular).
9.3 Muestras de
control de calidad (MCC).
9.3.1 Muestras de
evaluación del desempeño.- Incluye las muestras en medio de todos lo lotes que
pueden muestrearse (ciegas o doble ciegas) conteniendo cantidades conocidas de
isómeros no marcados del 2,3,7,8-sustitutos PCDDs/PCDFs u otros isómeros
PPCDDS/PPCDFS.
9.3.2 Soluciones de
verificación del desempeño.
9.3.2.1 Al inicio de cada
periodo de 12 horas durante el cual las muestras serán analizadas se debe
analizar una alícuota de; 1) la
solución de verificación del desempeño de la columna GC, y 2) solución de calibración de concentración de alta resolución No.
3 (HRCC-3; ver Tabla 5); con el fin de demostrar la resolución del GC adecuada
y la sensibilidad, la reproducibilidad de los factores de respuesta, el
intervalo de calibración y establecer los intervalos del tiempo de retención
del PPCDDS/PPCDFS. Se debe verificar una resolución de masas y el desempeño
para demostrar una resolución de masa adecuado usando un compuesto de
referencia apropiado (se recomienda PFK). Si no se alcanza el criterio
requerido, deben tomarse acciones correctivas antes de analizar cualquier
muestra.
9.3.2.2 Para validar
positivamente los datos de la muestra, la rutina o la calibración continua (HRCC-3;
Tabla 5) y la resolución de la masa debe verificarse el desempeño también al
final de cada periodo de 12 horas durante el cual las muestras son analizadas.
Además, un blanco de reactivos corrido en el HRGC/HRMS debe registrarse
siguiendo una corrida de calibración y la primera muestra corrida.
a) Si el laboratorio opera sólo
durante un periodo cada día de 12 horas o menos, la solución de verificación
del desempeño del GC debe analizarse sólo una vez (al inicio del periodo) para
validar los datos adquiridos durante el periodo. Sin embargo, la resolución de
masas y la verificación de la calibración continua debe desarrollarse al inicio
además de al final del periodo.
b) Si
el laboratorio opera durante periodos de 12 horas consecutivos, el análisis de la
solución de verificación del desempeño del GC debe desarrollarse al inicio de
cada periodo de 12 horas. La resolución de masas y la verificación de la
calibración continua de los periodos previos puede usarse para el inicio del
siguiente periodo.
9.3.2.3 Los resultados de
al menos uno de los análisis de la solución de verificación del desempeño de la
columna GC y de dos resoluciones de masa y la verificación de la calibración
continua debe reportarse con el dato de la muestra obtenido durante el periodo
de 12 horas.
9.3.2.4 Las desviaciones
de los criterios especificados para la verificación del desempeño del GC o la
verificación para la resolución del masas, invalida todos los datos obtenidos
de las muestras positivas entre análisis de la solución de verificación del
desempeño y los extractos de aquellas muestras positivas, por lo tanto deberán
volverse a analizar. Si la rutina de la corrida de calibración falla al inicio
de las 12 horas, deben seguirse las instrucciones de la sección de calibración.
Si falla la verificación de la calibración continua al final del periodo de 12
horas en no más del 25% de RPD para los 17 compuestos no marcados y 35% del RPD
para los 9 compuestos de referencia marcados, usar la media del RRFs de las dos
rutinas de calibración diaria, calcular la concentración del analito, en lugar
de obtener el RRFs obtenido de la calibración inicial. Una nueva calibración
inicial (nuevo RRFs) se requiere inmediatamente (dentro de dos horas) seguido
por el análisis de muestras, siempre que el RPD de la rutina de calibración
final exceda el 25% o el 35%, respectivamente. Una falla en el desempeño lleva
necesariamente a una nueva calibración inicial seguido inmediatamente del
análisis de las muestras esto conlleva a reanalizar todos los extractos de las
muestras positivas analizadas antes de que fallará la verificación de la
calibración continua final.
9.3.3 La mezcla de la
verificación del desempeño del GC, las soluciones de calibración de
concentración de alta resolución, y la solución de fortificación de la muestra
puede obtenerse del EMSL-CIN. Sin embargo, si no se dispone del EMSL-CIN, los
estándares deben obtenerse de otras fuentes, y las soluciones pueden prepararse
en el laboratorio. Las concentraciones de todas las soluciones conteniendo
2,3,7,8-sustituto PCDDs/PCDFs, los cuales no se obtienen del EMSL-CIN, deben
verificarse por comparación con las soluciones estándares certificadas que
están disponibles del EMSL-CIN.
9.3.4 Blancos de
campo.- Cada lote de muestras contienen una muestra de blanco de campo que será
fortificado antes del análisis de acuerdo a la sección 9.3.4.1. En adición a este blanco de campo, un lote de muestras
puede incluir un enjuague, el cual es una porción del solvente (usualmente
tricloroetileno) que fue usado para enjuagar el equipo de muestreo. El enjuague
es analizado para asegurar que las muestras no fueron contaminadas por el
equipo de muestreo.
9.3.4.1 Blanco de campo
fortificado.
a) A un blanco de campo adicionar
100 µL de la solución que contiene los nueve estándares internos (Tabla 17.3) diluida con 1,0 ml de acetona.
b) Extraer
según el procedimiento de las secciones 11.3.1
u 11.3.2, según aplique, adicionar
10 µL de la solución del estándar de recuperación y analizar una alícuota de 2
µL del extracto concentrado.
c) Calcular la concentración del
2,3,7,8-sustituto del PCDDs/PCDFs (sección 12.2) y el porcentaje de recuperación de los estándares internos.
d) Extraer y analizar un nuevo
blanco de campo fortificado cada vez que un nuevo lote de solventes o reactivos
sean utilizados para la extracción de la muestra o para procedimientos de
columnas cromatográficas.
9.3.4.2 Muestra de
enjuague.
a) La
muestra de enjuague debe fortificarse como una muestra regular.
b) Tomar
una porción de 100 ml (± 0,5 ml) de la muestra de solvente de enjuague del
equipo (muestra de enjuague), si es necesario filtrar y adicionar 100 µL de la
solución conteniendo los nueve estándares internos (Tabla 17.3).
c) Usando
un aparato K-D, concentrar un volumen aproximado de 5 ml.
NOTA: Como una opción,
un evaporador rotatorio puede usarse en lugar del aparato K-D para la
concentración del enjuague.
d) Transferir los 5 ml
concentrados del tubo concentrador K-D en porciones de 1 ml a un minivial de
1-ml, reduciendo el volumen en el minivial si es necesario con vapor de
nitrógeno seco.
e) Enjuagar el tubo concentrador
K-D con dos porciones de 0,5 ml de hexano y transferir los enjuagues al
minivial de 1 ml. Aplicar nitrógeno seco tanto como sea necesario.
f) Justo
antes del análisis, adicionar 10 µL de la solución estándar de recuperación
(Tabla 17.3) y reducir el volumen
hasta su volumen final, tanto como sea necesario. No se requiere columna
cromatográfica.
g) Analizar
una alícuota siguiendo los mismos procedimientos para el análisis de muestras.
h) Reportar
el porcentaje de recuperación del estándar interno y la presencia de cualquier
compuesto PPCDDS/PPCDFS en µg/L del enjuague de solvente.
9.3.5 Análisis
Duplicado.- En cada lote de muestras, colocar la muestra específica para el
análisis duplicado y analizar una segunda porción de muestra.
9.3.5.1 Los resultados de
los duplicados (porcentaje de recuperación y concentración de los compuestos
2,3,7,8-sustituto de PPCDDS/PPCDFS) deben coincidir con el 25% de la diferencia
relativa (la diferencia se expresa como porcentaje de la media). Reportar todos
los resultados.
9.3.5.2 Acciones
recomendadas para ayudar en la detección de problemas.
a) Verificar satisfactoriamente el
desempeño del instrumento (secciones 9.2
y 9.3). Si es posible, verificar que
no se cometieron errores en el pretratamiento de la muestra.
b) Revisar el procedimiento
analítico con el desempeño del personal del laboratorio.
9.3.6 Matriz adicionada
(MA) y matriz adicionada duplicada (MAD).
9.3.6.1 Establecer la
muestra para el análisis de MA y MAD (la muestra puede marcarse “volumen
doble”).
9.3.6.2 Adicionar un
volumen apropiado de la solución de fortificación de la matriz adicionada
(sección 7.10) y de la solución de
la fortificación de la muestra (sección 7.8),
ajustar el nivel de fortificación como se especifica en la Tabla 17.2 bajo niveles de adición IS.
9.3.6.3 Analizar las
muestras MA y MAD como se describe en la sección 11.
9.3.6.4 Los resultados
obtenidos de las muestras MA y MAD (concentraciones de 2,3,7,8-sustituto
PCDDs/PCDFs) deben concordar dentro de 20% de la diferencia relativa.
9.4
Estándares Internos del Porcentaje de Recuperación.- Para cada muestra,
calcular el porcentaje de recuperación del blanco de reactivos y el enjuague.
El porcentaje de recuperación debe estar entre el 40 y el 135% para todos los
estándares internos del 2,3,7,8-sustituto.
NOTA: Un
porcentaje de recuperación alto o bajo para un blanco no requiere descartar el
dato analítico pero puede indicar un problema potencial con futuros datos
analíticos.
9.5
Criterios de identificación.
9.5.1 Si
cualquiera de los criterios de identificación aparecen en las secciones 11.5.4.1(a) hasta la 11.5.4.1(d) y no son cumplidos para una
serie de homólogos, se reporta que la muestra no contiene un isómero marcado
del 2,3,7,8-sustituto PPCDDS/PPCDFS para aquellas series de homólogos al
calcular el límite de detección.
9.5.2 Si
el primer criterio de identificación inicial (secciones 11.5.4.1(a) hasta la 11.5.4.1(d))
se cumple, pero los criterios que aparecen en las secciones 11.5.4.1(e) y 11.5.4.2(a) no se cumplen, se presume que aquellas muestras
contienen contaminantes que interfieren. Esto debe anotarse en la forma de
reporte analítico y la muestra debe volver a correrse o reanalizarse el
extracto.
9.6
Porciones no utilizadas de muestras o extractos de muestras deben preservarse
durante seis meses después de entregados los resultados.
9.7 Debe
minimizarse el reuso de material para evitar problemas de contaminación.
10.
Calibración
10.1
Calibración inicial.- La calibración inicial se requiere antes del análisis de
cualquier muestra para PCDDs y PCDFs. También si cualquier rutina de
calibración no cumple con los criterios.
10.1.1 Las
cinco soluciones de calibración de concentración de alta resolución listadas en
la Tabla 17.6 deben usarse para la
calibración inicial.
10.1.2
Preparar el instrumento con PFK como se describe en la sección 11.4.2.1.
10.1.3
Inyectar 2 µL de la solución de verificación del desempeño de la columna GC
(sección 7.7) y obtener los datos de
la masa espectral SIM como se describe en la sección 11.4.2.2. El ciclo de tiempo total debe ser < 1 seg. El
laboratorio no debe desarrollar cualquier análisis extra hasta que se demuestre
y documente que los criterios listados en la sección 9.2.1. se han cumplido.
10.1.4
Usando las mismas condiciones de GC y MS que producen resultados aceptables con
la solución de verificación del desempeño de la columna, analizar 2 µL de la
porción de cada una de las cinco soluciones de calibración concentradas con los
siguientes parámetros de operación del espectrómetro de masas.
10.1.4.1 La
proporción de la corriente del integrador de iones para los iones que aparecen
en la Tabla 17.9 (series de
homólogos de la cuantificación de iones) debe estar dentro de los límites de
control indicados (grupo para cada serie de homólogos).
10.1.4.2 La
proporción de la corriente del integrador de iones para los iones
pertenecientes a los estándares internos y de recuperación con carbones
marcados deben estar dentro de los límites de control estipulados en la Tabla 17.9.
NOTA: Las
secciones 10.1.4.1 y 10.1.4.2 requieren que las 17
proporciones de los iones de la sección 10.1.4.1
y las once proporciones de los iones de la sección 10.1.4.2 estén dentro de los límites especificados de control
simultáneamente en una corrida. Es la responsabilidad del laboratorio tomar
acciones correctivas si la proporción de las abundancias de los iones están
fuera de los límites.
10.1.4.3 Para
cada SICP y para cada señal GC correspondiendo a la elución de los analitos
marcados y de sus estándares marcados, la proporción señal-a-ruido (S/N) debe
ser mejor que o igual a 2,5.
Se requiere la medición de S/N para cualquier
pico GC que tiene un S/N aparente de menos de 5:1. El resultado de los cálculos debe aparecer en el SICP por arriba
del pico GC en cuestión.
10.1.4.4 Con
referencia en la Tabla 17.10,
calcular los 17 factores de respuesta relativos (RRF) para los analitos no
marcados [RRF(n); n = 1 a 17] relativo a sus estándares internos apropiados
(Tabla 17.6) y los nueve RRFs para
los estándares internos marcados 13C12 [RRF(m); m = 18 a 26] relativo a los dos
estándares de recuperación de acuerdo a la siguiente ecuación:
Ecuación 2:
RRF(n) = A(x) * Q(is)/Q(x) * A(is)
Ecuación 3:
RRF(m) = A(is) * Q(rs)/Q(is) * A(rs)
donde:
A(x) = suma de las
abundancias de la integración de iones de la cuantificación de iones (Tablas 17.7 y
17.10) para PCDDs/PCDFs
marcados.
A(is) = suma de las abundancias
de los iones integrados de los iones cuantificados (Tablas 17.1 y 17.10) para los estándares
internos marcados.
A(rs) = suma de las abundancias
de la integración de iones de los iones cuantificados (Tablas 17.7 y
17.10) para los estándares de
recuperación marcados.
Q(is) = cuantificación del estándar
interno inyectado (pg).
Q(rs) = cuantificación de los
estándares de recuperación inyectados (pg), y
Q(x) = cuantificación de los
analitos PPCDDS/PPCDFS no marcados inyectados.
El RRF(n) y el RRF(m) son cantidades
adimensionales; las unidades usadas para expresar Q(is), Q(rs) y Q(x) deben ser
las mismas.
10.1.4.5
Calcular el RRF y sus respectivos porcentajes de desviación relativa estándar
(%RSD) para las cinco soluciones de calibración:
Ecuación 4:
RRF(n) = 1/5 RRFj (n)
j = 1
donde:
n = representa un congénere
particular (2,3,7,8-sustituto) del PPCDDS/PPCDFS (n = 1 a 17; Tabla 17.10);
j = es el número de inyección
(o número de solución de calibración; j = 1 a 5), y
10.1.4.6 Los
factores de respuesta relativos que serán usados para la determinación de la
concentración de los isómeros totales en las series de homólogos (Tabla 17.10) son calculados como sigue:
a) Para isómeros que pertenecen a una serie de
homólogos que contienen sólo un isómero (por ejemplo, OCDD y OCDF) o sólo un
isómero 2,3,7,8-sustituto (Tabla 17.5;
TCDD, PeCDD, HPPCDDs y TCDF), la media del RRF usado será la misma que la media
del RRF determinado en la sección 10.1.4.5.
NOTA: Las
soluciones de calibración no contienen estándares internos tal como 13C12-OCDF. Esto es
debido a un poder de resolución de 12,000 se requiere para resolver el ión
[M+6](+) del 13C12-OCDF del ión
[M+2](+) de OCDD (y [M+4] (+) de 13C12-OCDF con [M] (+) de OCDD). Por lo tanto, el
RRF para el OCDF se calcula relativo a 13C12-OCDD.
b) Para isómeros que pertenecen a una serie de
homólogos con más de un isómero 2,3,7,8-sustituto (Tabla 17.5), la media del RRF usado para aquella serie de homólogos será
la media calculada del RRFs para todos los isómeros 2,3,7,8-sustitutos usando la
siguiente ecuación:
Ecuación 5:
RRF(k) = 1/t RRF
n = 1
donde:
k = 27 a 30 (Tabla 17.10), con 27 = PeCDF; 28 = HxCDF; 29 =
HxCDD y 30 = HPPCDFs,
t = número total de los
isómeros 2,3,7,8-sustitutos presentes en las soluciones de calibración (por
ejemplo, dos para PeCDF, cuatro para HxCDF, tres para HxCDD y dos para
HPPCDFs).
NOTA:
Presumiblemente, los factores de respuesta del HRGC/HRMS de diferentes isómeros
dentro de una serie de homólogos son diferentes. Sin embargo, este protocolo
analítico hará la suposición que la respuesta HRGC/HRMS de todos los isómeros
en una serie de homólogos que no tienen el patrón del 2,3,7,8-sustituto son el
mismo como las respuestas de uno o más de los isómeros 2,3,7,8-sustituto en
aquella serie de homólogos.
c) Los factores de respuesta relativos [RRF(m)]
que serán usados para la determinación de los porcentajes de recuperación para
los nueve estándares internos se calcula como sigue:
Ecuación 6:
RRF(m) = A(is)(m)*Q(rs)/Q(is)(m) *A(rs) 5
Ecuación
7:
RRF(m) = 1/5 RRFj(m)
j = 1
donde:
m = 18 a 26 (tipo de congénere);
j = 1 a 5 (número de inyecciones);
A(is)(m) = suma
de la abundancia de la integración de iones de los iones cuantificados (Tabla 17.7 y 17.10) para un estándar interno dado (m = 18 a 26);
A(rs) = suma de la abundancia de la integración de iones (Tabla 17.7 y 17.10) para los estándares de recuperación apropiados (ver Tabla 17.6, pies de nota);
Q(rs),
Q(is)(m) = 5 cantidades respectivamente, el estándar de recuperación (rs) y
un estándar interno particular (es 5 m) inyectado (pg);
RRF(m) = factor
de respuesta relativa de un estándar interno particular (m) relativo a un
estándar de recuperación apropiado, determinado de inyección, y
RRFj(m) = 5
factores de respuesta relativo promedio calculado de un estándar interno
particular (m) relativo a un estándar de recuperación apropiado, determinado de
las cinco inyecciones de la calibración inicial (j).
10.2 Criterios de
aceptación de la calibración- Los criterios listados abajo para la aceptación
de la calibración deben cumplirse antes del desarrollo del análisis.
10.2.1 El porcentaje de
la desviación estándar relativa para la media de los factores de respuesta
[RRF(n) y RRF(m)] de los 17 estándares no marcados no debe exceder ± 20% y
aquel para los 9 compuestos de referencia marcados no debe exceder ± 30%.
10.2.2 La relación S/N
para las señales GC presentes en cada SICP (incluyendo los primeros para los
estándares marcados) debe ser > 10.
10.2.3 Las proporciones
isotópicas (Tabla 17.9) deben estar
dentro de los límites de control especificados.
NOTA: Si los criterios
de aceptación de la calibración listados en la sección 10.2.1 se cumplen, el RRF analítico especificado puede entonces ser
considerado como independiente de la cantidad del analito para el intervalo de
concentración de la calibración. La media del RRF será utilizado para todos los
cálculos hasta que la rutina de calibración no cumpla los criterios. En ese
momento, debe calcularse una nueva media del RRF a partir de un nuevo grupo de
inyecciones de las soluciones de calibración.
10.3 Rutina de
Calibración (Verificación de la Calibración Continua).- Las rutinas de
calibración deben desarrollarse al inicio de cada periodo de 12 horas después de
verificar un desempeño exitoso de la resolución de masas y la resolución del
GC. Una rutina de calibración también se requiere al final de un periodo de 12
horas.
10.3.1 Inyectar 2 µL del
estándar HRCC-3 de la solución de calibración concentrada (Tabla 17.6). Usando las mismas condiciones del
HRGC/HRMS de la sección 11.4.1 y 11.4.2, determinar y documentar una
calibración aceptable como se provee en la sección 10.4.
10.4 Criterios para la
Aceptación de la Rutina de Calibración-Los siguientes criterios deben cumplirse
antes de que se desarrolle el resto del análisis.
10.4.1 Los RRFs medidos
[RRF(n) para los estándares no marcados] obtenidos durante las corridas de la
rutina de calibración deben estar dentro de ± 20% de los valores medios
establecidos durante la calibración inicial (sección 10.1.4.5).
10.4.2 Los RRFs medidos
[RRF(m) para los estándares marcados] obtenidos durante las corridas de la
rutina de calibración debe estar dentro ± 30% de los valores medios
establecidos durante la calibración inicial (sección 10.1.4.7).
10.4.3 La proporción de
la abundancia de iones (Tabla 17.9)
debe estar dentro de los límites de control permitidos.
10.4.4 Si cualquiera de
los criterios de las secciones 10.4.1
y 10.4.2 no se satisfacen, repetir
una vez más. Si estos criterios no sean satisfecho aún, el proceso de rutina de
calibración completo debe revisarse. Esto se realiza ya que no siempre se puede
guardar todos los criterios de RRF. Por ejemplo, ha ocurrido que el criterio
del RRF para 13C12-HPPCDDs y 13C12-OCDD no se
cumple; sin embargo, los valores de RRF para los compuestos correspondientes no
marcados estuvieron rutinariamente dentro de los criterios establecidos en el
método. En estos casos, 24 de los 26 parámetros RRF tienen que cumplir con los
criterios de control de calidad de los datos para los valores de los HPPCDDs y
OCDD no marcados, no fueron acordados como un resultado del evento de
calibración. En estas situaciones, el analista debe registrar el efecto que
cubre todos los datos como se requiere para los datos de calidad y decidir una
acción apropiada. Deben tomarse acciones correctivas en razón, por ejemplo, si
los compuestos para los cuales los criterios RRF no fueron cumplidos incluyendo
tanto los estándares internos correspondiente como los compuestos no marcados.
Si los criterios de la proporción de la abundancia-ión no se satisfacen,
refiera a la nota de la sección 10.1.4.2
para la resolución.
NOTA: Una calibración
inicial debe llevarse a cabo siempre que el HRCC-3, la fortificación de la muestra
o la solución del estándar de recuperación se reemplaza por una nueva solución
de un lote diferente.
11.
Procedimiento
11.1
Muestreo.- Debido a la complejidad del método, se requiere que los
muestreadores estén entrenados y tengan amplia experiencia para asegurar la
veracidad y reproducibilidad de los resultados.
11.1.1
Pruebas preliminares obligatorias.- Todos los componentes deben calibrarse de
acuerdo a los procedimientos APTD-0576 o una calibración similar.
11.1.1.1
Pesar varias porciones de 200 a 300 g de sílica gel en contenedores
herméticos con una precisión de 0,5 g o como una alternativa pesar directamente
en los impactores que serán utilizados en el tren de muestreo un poco antes de
utilizarse.
11.1.1.2
Revisar los filtros visualmente a contraluz para detectar fallas en el filtro y
prevenir fugas. Etiquetar los frascos que contendrán los filtros y guardar en
ellos los filtros pesados.
11.1.1.3
Estabilizar los filtros a 20 ± 5,6°C a presión atmosférica durante 24 horas
pesando los filtros cada 6 horas para asegurarse que se ponen a peso constante
o sea que no deberán variar más de 0,1 mg de la última pesada. Durante las
pesadas nunca deberán exponerse los filtros por más de 2 minutos a una humedad
relativa mayor a 50%. Alternativamente los filtros pueden secarse en un horno a
105°C por 3 horas y guardarse en desecador.
11.1.2
Determinaciones preliminares.- Seleccionar el sitio de muestreo y el número de
puntos de muestreo de acuerdo al procedimiento descrito en el Método de
muestreo de emisiones en fuentes estacionarias para determinación de metales.
11.1.2.1
Determinar la presión de la chimenea, temperatura y velocidad de los gases, de
acuerdo al procedimiento descrito en el método descrito en la Norma Mexicana
NMX-AA- 009-93, verificar las fugas en las líneas del tubo Pitot.
11.1.2.2
Determinar el contenido de humedad utilizando el procedimiento descrito en el
método descrito en la Norma Mexicana NMX- AA-54-1978.
11.1.2.3
Determinar el peso molecular del gas de la chimenea en base seca de acuerdo al
procedimiento descrito en el Método de muestreo de emisiones en fuentes
estacionarias para determinación de metales.
11.1.2.4
Seleccionar el tamaño de la boquilla según la velocidad de los gases, de tal
manera que no sea necesario algún cambio de boquilla para poder mantener el
muestreo isocinético. Durante la corrida no cambiar el tamaño de la boquilla.
11.1.2.5
Asegurar que la presión diferencial sea adecuada y seleccionar el rango de
velocidades que se encontrarán en la chimenea durante la corrida.
11.1.2.6
Seleccionar la longitud adecuada de la sonda de muestreo, de manera que todos
los puntos transversales puedan muestrearse. Para muestrear chimeneas grandes
considerar el muestreo por los lados opuestos para poder determinar el número
suficiente de puntos transversales.
11.1.2.7 El
tiempo total de muestreo debe ser mayor o igual al tiempo mínimo para muestrear
el volumen requerido.
11.1.2.8 Se
recomienda que el tiempo en cada punto transversal muestreado sea al menos de
2,0 minutos.
11.1.3
Limpieza del Material.- Todas las partes de vidrio del tren de muestreo,
incluido el módulo de la resina y los impactores deben lavarse como se
especifica en la Sección 3 A del Manual de Métodos Analíticos para Análisis de
Residuos de Plaguicidas en Muestras Ambientales y Humanas de la USEPA. Debe
tenerse especial cuidado en la limpieza de las partes que tuvieron grasa de
silicona en las conexiones de vidrio. Los residuos de grasa deben removerse
completamente por el remojo durante varias horas en mezcla crómica antes de la
rutina de limpieza mencionada.
11.1.3.1 Todo
el material de vidrio debe enjuagarse con cloruro de metileno antes de usarse
en el tren de muestreo de PCDDs/PCDFs.
11.1.4
Módulo de la Resina XAD-2.- Usar una
cantidad suficiente (al menos 30 g o 5 gramos/m3 del gas a muestrearse) de resina XAD-2 limpia
para llenar completamente el módulo contenedor, el cual ha sido previamente
limpiado y enjuagado con hexano. La trampa y la lana de vidrio deben enjuagarse
en repetidas ocasiones con los disolventes que se utilizan para lavar la
resina. El contenedor de la resina no debe destaparse hasta que se va a montar
el tren de muestreo.
11.1.4.1 La
adición de los estándares surrogados debe realizarse en el laboratorio de 6 a
12 horas antes de iniciar el muestreo.
11.1.5 Si
se usan impactores para condensar la humedad de los gases de la chimenea
prepararlos de la siguiente manera:
11.1.5.1
Colocar 100 ml de agua en el primer impactor y 100 ml de etilenglicol o agua en
el segundo, en el tercer impactor colocar 250 a 300 gramos de sílica gel
previamente pesada, anotar en la bitácora el peso, ya que se utilizará para el
cálculo de la humedad por gravimetría.
NOTA: No
utilizar grasas de silicón o de otra clase para sellar fugas.
11.1.5.2
Colocar el contenedor en un lugar limpio para más tarde utilizarlo en la
recuperación de la muestra.
11.1.5.3
Utilizando pinzas o guantes colocar el filtro en el portafiltros asegurándose
que quede centrado de tal manera que el empaque quede sellado, revisar el portafiltros
y el filtro para detectar rasgaduras.
11.1.5.4
Marcar los puntos transversales a muestrear con cinta resistente al calor
colocándola alrededor de la sonda.
11.1.5.5
Ensamblar el tren de muestreo y colocar hielo en trozos alrededor de los
impactores.
11.1.6
Procedimiento para revisión de fugas.
11.1.6.1
Prueba previa de revisión de fugas. Se requiere una verificación previa de
fugas según el siguiente procedimiento:
11.1.6.2
Después de que el tren de muestreo ha sido ensamblado encender el sistema de
calentamiento de la sonda y esperar a que alcance las condiciones requeridas de
operación, permitir que la temperatura se estabilice. Revisar fugas en todas
las uniones y conexiones del tren de muestreo y conexiones de la boquilla por
medio de la hendidura de la boquilla con un tapón de PTFE limpio, aplicar vacío
de 380 mm de Hg. (15”).
NOTA:
Puede usarse un vacío más bajo a condición de que en ningún momento de la
prueba se rebase tal vacío.
11.1.6.3 Una
fuga en exceso de 4% del promedio del rango de muestreo o 0,00057 metros
cúbicos por minuto es inaceptable.
11.1.7
Verificación de fugas en el tren de muestreo.- Para verificar fugas en el tren
de muestreo, utilizar el siguiente procedimiento:
11.1.7.1
Iniciar bombeando con la válvula de “by-pass” totalmente abierta y la válvula
del ajuste grueso totalmente cerrada. Parcialmente abrir la válvula del ajuste
grueso y lentamente cerrar la de “by-pass” hasta obtener el vacío adecuado.
No regresar la válvula del “by-pass” ya que
podría regresar agua al portafiltro, si el vacío deseado se excede, verificar
las fugas a ese valor de vacío y terminar de revisar las fugas como se indica
abajo y continuar.
11.1.7.2
Cuando se termina de revisar las fugas, primero remover lentamente el tapón que
se colocó en la boquilla, inmediatamente apagar la bomba de vacío. Debe
cuidarse que la sílica gel no se humedezca.
11.1.8
Verificación de fugas durante el muestreo:
11.1.8.1 Si
durante el muestreo un componente (ejemplo el filtro, un impactor u otro
ensamble) se cambia, deben verificarse las fugas inmediatamente después de
haber hecho el cambio. La verificación de las fugas debe realizarse según la
sección 11.1.6 excepto que deberá
hacerse a un vacío igual o mayor al valor máximo registrado en la prueba. Si la
fuga detectada presenta un flujo menor a 0,00057 m3/min o 4% del
flujo promedio muestreado el resultado es aceptable sin necesidad de
corrección. Si la fuga es mayor a lo estipulado corregir el volumen total según
la sección 11.1.10 de este método o
el muestreo se debe anular.
11.1.9
Verificación de fugas posterior.
11.1.9.1 Es
obligatorio una verificación de fugas al concluir cada uno de los muestreos. La
verificación de fugas deberá realizarse según el procedimiento de la sección 11.1.6 excepto que debe conducirse a un
vacío igual o mayor al máximo registrado durante la prueba. Si el flujo de la
fuga no es mayor a 0,00057 m3/min o 4% del promedio del flujo muestreado el
resultado es aceptable y no requiere que se aplique la corrección al total del
flujo muestreado. Si la fuga es mayor corregir el volumen final según la
sección 11.3 de este método o el
muestreo se debe anular.
11.1.10
Corrección de una fuga excesiva.
11.1.10.1 El
volumen muestreado obtenido mediante la ecuación que se da en la sección 12.3 de este método, se corrige por
medio de la ecuación 5 si excede el máximo aceptable de fugas, reemplazar (Vm)
en la ecuación 12.3 por:
Ecuación 8:
Vm = (Li-La)Oi-(Lp-La)Op
donde:
Vm = Volumen de gas muestreado y
medido.
La = Máximo aceptable de fugas
0,00057 metros cúbicos por minuto (0,02 cfm) o 4% del flujo promedio
muestreado.
Lp = Volumen de fuga observada
durante la verificación de fugas al final de la prueba.
Li = Volumen de fuga
observado durante la verificación realizada en los diferentes muestreos
(i=1,2,3,...n), metros cúbicos por minuto.
Oi = Tiempo de muestreo entre
dos sucesivas verificaciones de fugas, iniciando con el intervalo de primero y
el segundo.
Op = Tiempo de muestreo entre
la última verificación de fugas y la prueba final.
Substituya solamente los siguientes rangos (Li
o Lp) los cuales excedan (La).
11.1.11
Operación del Tren.
11.1.11.1
Durante el muestreo, mantener el flujo dentro de un 10% del valor
isocinético real; para cada corrida registrar la información requerida en la
hoja de campo. Asegúrese de registrar la lectura inicial del equipo. Registrar
las lecturas del equipo al principio y al final de cada incremento de tiempo en
el muestreo, cuando se realice un cambio en el flujo, antes y después de cada
verificación de flujo y cuando se suspenda el muestreo.
11.1.11.2
Registrar otras lecturas requeridas en la hoja de campo y llenar una por cada
punto de muestreo transversal, por cada incremento significativo que tenga un
20% de variación en la velocidad, se requieren ajustes adicionales en los
flujos.
11.1.11.3
Nivelar y ajustar a cero el manómetro inclinado.- El nivel y el cero del
manómetro pueden tener variaciones debido a vibraciones y variaciones de
temperatura, realizar verificaciones periódicas.
11.1.11.4
Limpiar los puertos de muestreo antes de la corrida de prueba para minimizar el
muestreo de los depósitos de material. Para iniciar el muestreo remover la
boquilla y verificar que el tubo Pitot y la extensión de la sonda sea la
adecuada y que se encuentren bien colocadas. Colocar la boquilla en el primer
punto transversal con la punta directamente hacia el flujo de gas.
11.1.11.5
Inmediatamente iniciar el bombeo y ajustar el flujo a la condición isocinética.
Si se disponen de nomogramas, éstos sirven de ayuda para ajustar rápidamente el
muestreo isocinético.
11.1.11.6
Estos nomogramas están diseñados para cuando se usan tubos Pitot tipo “S” con
un coeficiente (Cp) de 0,85 ± 0,02 y la densidad del gas equivalente de la
chimenea (peso molecular promedio en base seca) (Md) sea igual a 29 ± 4. Si Cp y Md están fuera de los rangos
de inicio, no usar el nomograma sin que se tomen los pasos apropiados para
compensar las desviaciones.
11.1.11.7
Cuando las chimeneas tienen una presión negativa significativa, tener atención
al cerrar la válvula del ajuste grueso antes de insertar la sonda dentro de la
chimenea para prevenir que el agua se regrese. Si es necesario, la bomba puede
encenderse con la válvula del ajuste grueso cerrada, cuando la sonda esté en
posición, bloquear o tapar alrededor de la sonda en el puerto de muestreo para
que no haya dilución de los flujos de gases.
11.1.11.8
Cuando se esté muestreando la sección transversal de la chimenea debe tenerse
cuidado de no bombear cuando se está ensamblando la boquilla en la sonda para
no remover material sólido de las paredes.
11.1.11.9 Durante la
corrida de prueba tomar las precauciones necesarias para mantener la
temperatura del condensador por debajo de 20°C (colocar trozos de hielo al baño
de hielo del impactor). Esto ayuda a evitar la pérdida de humedad. También
revisar periódicamente el nivel y el cero del manómetro.
11.1.11.10 Si la caída de
presión aumenta en el filtro y esto impide la medición isocinética, reemplazar
el filtro durante la corrida. Es recomendable se tenga preparado todo un nuevo
ensamble o portafiltro para usarse. Antes de operar el nuevo ensamble realizar
una verificación de fugas. El total de las partículas será recolectado en los
dos filtros.
11.1.11.11 Debe utilizarse
un solo tren para obtener la muestra, excepto cuando se requieren muestreos
simultáneos de dos o más ductos separados o dos o más lugares diferentes del
mismo ducto.
11.1.11.12 Al final de la
corrida apagar la bomba, remover la sonda y registrar la lectura final del
equipo. Realizar una verificación de las fugas. Las líneas deberán pasar la
presente verificación de fugas para dar validez al trabajo.
11.1.12 Método de cálculo
del porcentaje de isocinetismo.- Calcular el % de isocinetismo para determinar si
la prueba fue válida, en caso contrario se debe llevar a cabo una segunda
prueba.
11.2 Recuperación de
la Muestra.
11.2.1 Al final del
muestreo es necesario limpiar la sonda tan rápido como sea posible para remover
la contaminación captada en la chimenea.
11.2.2 Cuando la
temperatura permita manejar la sonda con seguridad, limpiar todo el exterior,
remover la sonda del tren de muestreo y taparla con papel aluminio previamente
lavado con cloruro de metileno. Sellar la entrada del tren con su tapón y papel
aluminio enjuagado con hexano.
11.2.3 Transferir la
sonda y los impactores al área de recuperación previamente seleccionada, tapar
para asegurar que no se contamine ni se pierda la muestra. En el área de
recuperación de la muestra no se debe fumar.
11.2.4 Inspeccionar el
tren de muestreo e investigar si hay alguna anormalidad, por ejemplo, un filtro
roto, fuga de algún líquido, cambio de color, etc.
11.2.5 Contenedor No. 1.- Cuidadosamente remover el filtro
del portafiltros y colocarlo en el contenedor identificado como “Contenedor No.
1”. Utilizar unas pinzas previamente lavadas para manejar el filtro. Si es
necesario doblar el filtro, asegurándose que las partículas queden dentro del
mismo. Cuidadosamente transferir al contenedor las partículas y las fibras del
filtro que se hayan quedado en el contenedor del filtro. Para limpiar el
contenedor del filtro utilizar un cepillo seco e inerte.
11.2.6 Módulo de la
Resina.- Remover el módulo de la Resina XAD-2 del tren de muestreo y taparlo
con papel aluminio lavado con hexano.
11.2.7 Contenedor de
muestra No. 2.- Recuperar
cuantitativamente el material depositado en la boquilla, en la sonda, en la
línea de transferencia, en la parte frontal del portafiltro y en el ciclón, si
fue usado. Primero cepillar y luego enjuagar secuencialmente tres veces con
metanol, benceno y cloruro de metileno. Colocar todos los enjuagues en el
contenedor No. 2.
11.2.8 Contenedor de
muestra No. 3.- Enjuagar la parte
posterior del portafiltro, la conexión entre la línea y el refrigerante (si se
usó el condensador separado del contenedor de la resina) tres veces
secuencialmente con metanol, benceno y cloruro de metileno y colectarlos en el
contenedor No. 3. Si se usó una
trampa/condensador, el enjuague de la trampa debe realizarse en el laboratorio
después de remover la porción de resina XAD-2. Si se usó una trampa expulsora de agua, el contenido y los
enjuagues deben ponerse en el contenedor No. 3. Enjuagar tres veces con metanol, benceno y cloruro de metileno.
11.2.9 Contenedor de
muestra No. 4.- Remover el primer
impactor, secar y limpiar la parte exterior del impactor. El contenido y los
enjuagues depositarlos en el contenedor No. 4. Enjuagar el impactor secuencialmente tres veces con metanol,
benceno y cloruro de metileno.
11.2.10 Contenedor de
muestra No. 5.- Remover el segundo y
tercer impactor, secar la parte exterior y limpiar. Vaciar el contenido y los
enjuagues dentro del contenedor No. 5.
Enjuagar cada uno con agua tres veces.
11.3 Preliminares al
análisis.- En los muestreos de chimeneas van a resultar muestras líquidas y
sólidas para su análisis. Las muestras deben combinarse como sigue:
1. El
filtro y las partículas colectadas en el filtro (Contenedores No. 1 y No. 2).
2. El
contenedor de la muestra No. 3, la resina y los enjuagues del cartucho de
resina.
3. Contenedores
de la muestra No. 4 y No. 5.
Es
preferible que las muestras no se dividan para su análisis, ya que es muy
difícil obtenerlas homogéneas como generalmente ocurre con las muestras
líquidas. Las muestras sólidas tales como la resina no es homogénea y el filtro
es tan pequeño que el límite de detección probablemente no se alcance, si se
divide la muestra.
PRECAUCION:
Cuando se utiliza este método todas las operaciones deben realizarse en áreas
restringidas a personas que estén perfectamente entrenadas en las medidas de
seguridad y de protección para evitar exposiciones a la piel.
11.3.1 Extracción de
Muestras Líquidas.
11.3.1.1 Contenedor de
muestra No. 2.- Concentrar los
enjuagues del contenedor de muestra No. 2 a un volumen de 1 a 5 ml usando un
Turbo-Vap a una temperatura de 50°C. El residuo contendrá partículas que fueron
removidas del tren de muestreo. Combinar el residuo (enjuague tres veces con el
contenido del recipiente final de la muestra) en el Soxhlet con el filtro y las
partículas y proceder como se indica en la sección 11.3.2.1.
11.3.1.2 Contenedor de
muestra No. 3.- Concentrar los
enjuagues del contenedor de muestra No. 3 a un volumen de 1 a 5 ml usando un
Turbo-Vap a 50°C. Concentrar casi a sequedad. Combinar los residuos (con tres
enjuagues del recipiente de muestra final) en el Soxhlet con la muestra de
resina y proceder como se describe en la sección 11.3.2.1.
11.3.1.3 Contenedor No. 4
y No. 5.- Combinar el contenido del
frasco No. 4 y No. 5 en un embudo de separación. Extraer la muestra tres veces
con tres porciones de cloruro de metileno. Combinar la fracción orgánica en un
matraz que contenga Na2SO4 anhidro.
Adicionar 500 µL de tetradecano y concentrar a 500 µL en un Kuderna-Danish o
rotavapor y transferir el extracto a un tubo de prueba de 8 ml con hexano.
Combinar el extracto en el Soxhlet con las muestras sólidas como se describe en
la sección 11.3.2.1
11.3.2 Extracción de
Muestra Sólidas.
11.3.2.1 Filtro y
Partículas Retenidas.- El Soxhlet debe limpiarse por 8 horas mínimo con el
disolvente de extracción y el disolvente debe descartarse. Adicionar 20 gramos
de Na2SO4 en el cartucho.
Cortar el filtro en pequeños trozos y colocarlos en el lugar de la muestra
junto con los enjuagues (Secc. 11.3.1.1
a 11.3.1.3) sobre la parte alta del
sulfato de sodio anhidro, agregar la resina XAD-2, colocar un tapón de fibra de
vidrio lavada y adicionar 50 µL de los estándares internos isotópicamente
marcados. Colocar el cartucho en el Soxhlet, adicionar 250 ml de tolueno al
matraz.
11.3.2.2 Ensamblar el
Soxhlet, encender la parrilla de calentamiento y abrir la llave del agua del
refrigerante, dejar en reflujo durante 16 h. Después de la extracción, esperar
a que el Soxhlet se enfríe. Transferir a un matraz de 500 ml y adicionar
aproximadamente 500 µL de tetradecano. Adicionar aproximadamente 50 ml de
hexano y concentrar a un volumen de 500 µL en un Kuderna-Danish o Turbo-Vap.
Transferir el extracto a un tubo de prueba de 8 ml con hexano, almacenar el
extracto para su limpieza en columna.
11.3.2.3 Limpieza opcional
preliminar.- Ciertas muestras que se encuentran muy sucias pueden requerir una
limpieza preliminar antes de ser analizadas. En tal caso, seguir el
procedimiento siguiente:
a) Lavar
el extracto orgánico con 25 ml de agua agitando 2 minutos, permitir reposar
para que se separen las fases, descartar la fase acuosa y la fase orgánica
transferirla a un matraz Erlenmeyer.
PRECAUCION:
Adicionar 50 ml de ácido sulfúrico concentrado al extracto orgánico, agitar por
10 minutos. Esperar que la mezcla se separe en un embudo de separación
(aproximado 10 min). Descartar con mucho cuidado la fase acuosa/ácida. Repetir
la operación hasta que el ácido casi no presente coloración.
b) Colocar la muestra en un embudo
de separación y adicionar 25 ml de agua, agitar 2 minutos y permitir separar
las fases. Descartar la capa acuosa y secar la capa orgánica con sulfato de
sodio anhidro.
c) Transferir el extracto orgánico a tubos de
Turbo-Vap y evaporar a 55°C casi a sequedad.
d) Reconstituir en hexano antes de proceder con la columna
cromatográfica.
11.3.3
Columnas de Limpieza.- El extracto obtenido como se describe en la sección
anterior es concentrado a 1 ml utilizando el Turbo-Vap. Transferir
cuantitativamente con tres enjuagues de 1 ml a la columna de sílica gel/alúmina
como se describe a continuación.
11.3.3.1
Preparación de columna combinada de sílica gel/alúmina.- Se empaca por gravedad
una columna de 200 mm por 15 mm, colocar lana de vidrio inerte (silanizada) en
la punta de la columna y adicionar en secuencia, 1 g de sílica gel, 2 g de
sílica gel modificada básica, 1 g de sílica gel, 4 g de sílica gel modificada
ácida, 1 g de sílica gel y una capa de 1 cm de sulfato de sodio anhidro.
11.3.3.2
Preparación de la columna ácida de alúmina.- Empacar por gravedad una columna
de vidrio de 11 mm de diámetro, colocar lana de vidrio inerte (Silanizada) en
la punta de la columna. Adicionar 6 g de alúmina ácida preparada como se
describe en la sección de reactivos, golpear suavemente la columna hasta que se
asiente la alúmina y adicionar 1 cm de sulfato de sodio anhidro.
11.3.3.3
Preparación de la columna de Carbopak-Celite.- Cortar a una pipeta serológica
de 5 ml 1 cm de la punta, colocar lana de vidrio silanizada y lavada con
cloruro de metileno. Adicionar suficiente Carbopak-Celite (0,3 g) a la columna
hasta hacer 2 cm de longitud y colocar un tapón de lana de vidrio en la parte
superior.
11.3.4
Procedimiento de Limpieza.
11.3.4.1
Eluir las columnas “A” y “B” con hexano y descartar el eluato. Revisar que la
columna no presente canales, si tiene descartarla. No tapar la columna húmeda.
11.3.4.2
Adicionar el extracto de muestra con 5 ml de hexano a la parte alta de la
columna “A” seguido de dos porciones de 5 ml de hexano para enjuagar. Eluir la
columna “A” con 90 ml de hexano directamente sobre la columna “B”. Eluir la
columna “B” con 20 ml de hexano/cloruro de metileno al 20% volumen. Concentrar
el extracto a 0,50 ml utilizando el Turbo-Vap.
NOTA: La
concentración óptima de cloruro de metileno va a variar con la actividad de la
alúmina. En cada lote de alúmina el analista debe determinar la concentración
óptima para eluir las bajas concentraciones de los estándares de calibración
sin eluir las interferencias de la columna.
11.3.4.3
Eluir la columna C con 5 ml de hexano en un sentido y luego en el sentido
inverso del flujo. Mientras está en el sentido inverso, eluir con 2 ml de
tolueno, 1 ml de cloruro de metileno/metanol/benceno (75/20/5) v/v, 1 ml de
cloruro de metileno/ciclohexano (50/50 v/v) y 2 ml de hexano. Descartar los
eluatos.
11.3.4.4
Mientras esté el flujo en la dirección inversa, transferir el concentrado de la
muestra a la columna con hexano y eluir la columna en secuencia con 1 ml de
hexano, 1 ml de cloruro de metileno/ciclohexano (50/50 v/v) y 1 ml de cloruro
de metileno/metanol/benceno (75/20/5 v/v). Descartar el eluato. Dé la vuelta a
la columna y eluir con 4 ml de tolueno. Almacenar este eluato para el análisis
de las PCDDs/PCDFs. Evaporar la fracción del tolueno a 1 ml aproximado en un
Turbo-Vap a 50°C.
11.3.4.5
Transferir a un microvial usando enjuagues de tolueno y concentrando a 50 µl
usando un flujo de nitrógeno extra seco. Almacenar los extractos en un
congelador protegidos de la luz hasta el momento del análisis por GC/MS.
NOTA: Los
extractos de las PCDDs/PCDFs son muy sensibles a la luz del sol, deben ser
guardados en viales ámbar y protegidos con papel aluminio de la luz en general,
bajas recuperaciones se obtendrán si no se protegen de la luz los extractos.
11.4
Condiciones y Adquisición de Datos del Cromatógrafo/Espectrómetro de Masas.
11.4.1
Cromatógrafo de Gases.
Cobertura de la
Columna: DB-5
Espesor de la
Capa: 0,25 µm
Dimensión de la
Columna: 60 m * 0,2 mm
Temperatura del
Inyector: 270ºC
Tiempo de Apertura
de la válvula: 45 seg.
Temperatura de
Interfase: Temperatura final
de la función.
Programa de
Temperatura:
|
Etapa |
Temperatura
Inicial (ºC) |
Tiempo
de Retención Inicial (min.) |
Temperatura
de Rampa (ºC/min.) |
Temperatura
Final (ºC) |
Tiempo
de Retención Final (min.) |
|
1 |
200 |
2 |
5 |
220 |
16 |
|
2 |
|
|
5 |
235 |
7 |
|
3 |
|
|
5 |
330 |
5 |
Tiempo total: 60 min.
11.4.2
Espectrómetro de Masas.
11.4.2.1 El
espectrómetro de masas debe operarse en modo de monitoreo de ión selectivo
(SIM) con un ciclo de tiempo total (incluyendo el tiempo de reseteo del
voltaje) de un segundo o menos (sección 11.4.3.1).
Como mínimo, deben monitorearse los iones listados en la tabla 17.7 para cada uno de los cinco SIM
descriptores. Note que con excepción del último descriptor (OCDD/OCDF9, todos
los descriptores contienen 10 iones. La selección (Tabla 17.7) de los iones moleculares M y M+2 del 13C12-HxCDF y 13C12-HPPCDFs además
del M+2 y M+4 (por consistencia) fue hecha para eliminar, aun bajo las
condiciones del espectrómetro de masas de alta resolución, las interferencias
que están ocurriendo en estos dos canales de iones en las muestras que
contienen altos niveles de HxPCDDs y HPPCDDss nativos. Es importante mantener
el mismo grupo de iones tanto para la calibración como para el análisis de los
extractos de las muestras. La selección del ión más cercano se permite para el
desarrollo del laboratorio.
NOTA: Como
una opción para el analista, las dioxinas y furanos tetra- y pentaclorados
pueden combinarse en un descriptor único.
11.4.2.2 Las
condiciones de tono recomendadas para el espectrómetro de masas están basadas
en el monitoreo de los grupos de iones mostrados en la Tabla 17.7. Usando una fuga del PKF molecular,
el tono del instrumento debe cumplir los requisitos mínimos del poder de
resolución de 10,000 (valle de 10%) a m/z 304.9824 (PKF) o cualquier otra señal de referencia cercana a m/z 303.9016 (del TCDF). Usando las mismas
condiciones del pico y el afore mencionado del pico de referencia, verifique
que la masa exacta de m/z 380.9760
(PKF) está dentro de 5 ppm del valor requerido. Note que la selección de los
iones de masa baja y alta deben ser tales que ellos proveen el salto de voltaje
más largo desempeñado en cualquiera de los cinco descriptores (Tabla 17.7).
11.4.3
Adquisición de datos.
11.4.3.1 El
ciclo total de tiempo para la adquisición de datos debe ser < 1 Seg. El
ciclo total de tiempo incluye la suma de todos los tiempos espaciados t los
tiempos del reseteo del voltaje.
11.4.3.2 Los
datos adquiridos del SIM para todos los iones listados en los cinco
descriptores de la Tabla 17.7.
11.5
Análisis.
11.5.1
Remover el extracto de la muestra o blanco del almacenamiento. Con un vapor de
secado, de nitrógeno purificado, reducir el volumen del extracto entre 10 y 50
µL.
NOTA: Un
volumen final de 20 µL o más debe usarse cuando sea posible. Aun el volumen
final de 10 µL es difícil de manejar y una inyección de 2 µL sacados de una
muestra pequeña de 10 µL para confirmación y repetición de inyecciones y para
guardar no es suficiente.
11.5.2
Inyectar una alícuota de 2 µL del extracto en el GC, operado bajo las
condiciones establecidas para producir resultados aceptables con la solución de
verificación del desempeño.
11.5.3
Obtenga los datos SIM de acuerdo a la sección 11.4.2 y 11.4.3. Usar las
mismas condiciones de adquisición de datos y de operación del espectrómetro de
masas previamente utilizadas para determinar los factores de respuesta
relativos (sección 10). Los iones característicos para los éteres bifenilos
policlorados están incluidos en los descriptores listados en la Tabla 17.7.
NOTA: El
periodo de adquisición debe al menos abarcar el intervalo del tiempo de
retención sobre todo PPCDDS/PPCDFS previamente determinado (sección 9.1). Los perfiles de las corrientes de
los iones selectivos (SICP) para los iones más cercanos (uno por descriptor de
masas) deben también registrarse e incluirse en el paquete de datos. Esos SICPs
deben ser representaciones verdaderas de la evolución de las amplitudes de los
iones más cercanos durante las corridas HRGC/HRMS. El analista puede ser
requerido para monitorear un ión PKF, no como la masa más cercana, pero como un
ión regular, en razón de cumplir estos requerimientos. Es recomendable para
examinar la masa más cercana del ión SICP para sensibilidades obviamente bajas
y cambios en la estabilidad del instrumento durante la corrida GC/MS que puede
afectar las medidas. Reporte cualquier discrepancia en el caso narrativo.
11.5.4
Criterios de Identificación.- Para un pico de cromatógrafo de gases que será
identificado como un PPCDDS o PPCDFS, deben cumplirse todos los siguientes
criterios:
11.5.4.1
Tiempos de Retención.
a) Para los isómeros 2,3,7,8-sustitutos, los
cuales tienen un estándar interno isotópicamente marcados o estándar de
recuperación presente en el extracto de la muestra (éste represente un total de
10 isómeros incluyendo el OCDD; Tablas 17.3
y 17.4), el tiempo de retención
(RRT; a un pico de máxima altura) de los componentes de la muestra (por
ejemplo, los dos iones usados para propósitos de cuantificación listados en la
Tabla 17.7) debe estar dentro de -1
a +3 segundos de los estándares isotópicamente marcados.
b) Para
los compuestos 2,3,7,8-sustitutos que no tienen estándares internos
isotópicamente marcados presentes en el extracto de la muestra (esto representa
un total de 6 isómeros; Tabla 17.4),
el tiempo de retención debe caer dentro de 0,005 unidades de tiempo de
retención del intervalo de tiempo de retención medido en la rutina de
calibración. La identificación del OCDD se basa en su tiempo de retención
relativo al 13C12-OCDD para
determinar desde los resultados de la rutina de calibración diaria.
c) Para compuestos
no-2,3,7,8-sustitutos (de tetra a octa; totalizando 119 isómeros), el tiempo de
retención debe estar dentro de los intervalos de tiempo de retención de los
homólogos correspondientes establecidos por el análisis de la solución de
verificación del desempeño de la columna (sección 9).
d) Las respuestas de la corriente
de iones tanto para los iones usados para propósitos de cuantificación (por
ejemplo, para TPCDDs: m/z 319,8965 y 321,8936) debe alcanzar un máximo
simultáneo (± 2 segundos).
e) Las
respuestas de la corriente de los iones, para tanto, los iones usados para los
estándares marcados (por ejemplo, para 13C12-TCDD:
m/z 331,9368 y m/z 333.9339) debe
alcanzar un máximo simultáneamente (± 2 segundos).
NOTA: Se requiere que
el analista verifique la presencia de 1,2,8,9-TCDD y 1,3,4,6,8,-PeCDF (sección
9) en el SICPs de la verificación del desempeño diariamente. En caso de que se
pierda un compuesto, el analista debe tomar acciones correctivas que indique la
habilidad para generar resultados de PPCDDSs/PPCDFSs.
f) Proporción
de la Abundancia de Iones- La corriente de los iones integrados para los dos
iones usados para propósitos de cuantificación, debe tenerse una proporción
entre los límites más bajos y más altos establecidos para las series de
homólogos, para los cuales los picos están asignados (Tabla 17.9).
g) Proporción
Señal/Ruido-Todas las intensidades de las corrientes de iones debe ser >
-2,5 veces el nivel de ruido para la identificación positiva de un compuesto
PPCDDS/PPCDFS o un grupo de isómeros coeluidos.
h) Interferencias
de Eteres Bifenilos Policlorados- En adición de los criterios de arriba, la
identificación de los picos GC como un PPCDFS puede sólo hacerse si la señal
teniendo un S/N > 2,5 es detectado, al mismo tiempo de retención (± 2
segundos), en el éter bifenilo policlorado (PCDPE, Tabla 17.7) canal.
12. Cálculos
Realizar
todos los cálculos al menos con una cifra decimal más de los datos obtenidos.
Redondear los cálculos en el resultado final.
12.1 Nomenclatura
An = Area seccional Cruzada de la boquilla (m2).
Bws = Vapor de agua en el gas, proporción por
volumen.
Cs = Concentración de PCDDs/PCDFs en gases de
chimenea, ng/dscm, corregida a condiciones estándar de 20°C, 760 mm Hg (68°F, 29.92 in Hg) en base seca.
Gs = Masa Total de PCDDs/PCDFs en muestras de gas
en chimenea, en ng.
I = % de isocinetismo en el muestreo.
La = Rango máximo aceptable de fuga para cada una
de las pruebas previas de verificación de
fugas o para la verificación de fugas seguido del cambio de un
componente; equivalente a 0,00057 m3/min o 4% del promedio del
rango de muestreo, cualquiera que sea el menor.
Li = El rango de fugas observado durante la
verificación de fugas realizado antes del tercer cambio de componente (I=1,2,3,....n) m3/min.
Lp = El rango de fugas observado durante la
verificación posterior a la toma de muestra.
M = Peso molecular del agua, 18,0 g/g-mole.
Pbar = Presión barométrica del sitio de muestreo, mm
Hg.
Ps = Presión absoluta de la chimenea, mm Hg.
Pstd = Presión estándar absoluta, 760 mm Hg.
R = Constante
ideal del gas 0,06236 mm Hg-m3 / °K -g-mol.
Tm = Promedio de la
temperatura absoluta del gas seco (°K).
NOTA: Tm
va a depender del tipo de medidor usado en la configuración del muestreo.
Ts = Promedio de la
temperatura absoluta del gas de la chimenea (°K).
T std = Temperatura estándar
absoluta (293°K).
Vaw = Volumen de acetona
utilizado en el lavado, ml.
V1c = Volumen total de líquido colectado en el
impactor y sílica gel, ml.
Vm = Volumen del gas
muestreado y medido con el equipo muestreador (m3).
Vm(std) = Volumen de gas muestreado
y medido por el equipo muestreador, corregido por las condiciones estándar (dsm3).
Vs = Velocidad del
gas de la chimenea, (m/seg).
Y = Factor de
calibración del medidor de gas seco.
DH = Promedio de la
presión diferencial del orificio transversal (mm H2O).
O = Tiempo total
muestreado en minutos.
O1 = Intervalo de tiempo muestreado, desde el inicio de la corrida hasta
el primer cambio de componentes, en minutos.
O! = Intervalo
de tiempo entre el cambio del primer componente y el segundo, en minutos.
Op = Intervalo
de tiempo, del cambio del componente final (nth) hasta el final de la corrida,
en minutos.
13.6 = Gravedad
específica del mercurio.
60 = seg/min.
100 = Conversión
a por ciento.
12.2
Promedio de temperatura del medidor de gas seco y promedio de la caída de
presión del orificio, ver los datos de la hoja de campo.
12.3
Volumen del gas seco.- Volumen de muestra corregido medido por el medidor de
gas seco a condiciones estándar (20°C, 760 mm Hg). Utilizando la ecuación 6.
Ecuación
9:
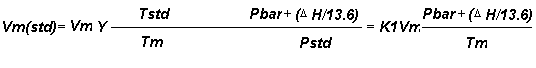
donde:
Ecuación
10:
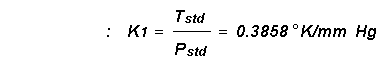
NOTA: La ecuación 6
puede ser usada como está escrita, si las fugas observadas durante alguna de
las verificaciones obligatorias (ejemplo: la verificación de fugas posterior a
la prueba o realizados por concepto de cambio de componentes) exceden La o si
Lp o L1 excede la ecuación 6 debe ser modificada como sigue:
(a) Caso No. 1 Si no
hay cambio de componentes durante la corrida de toma de muestra. En este caso
reemplace Vm en la ecuación 6 con la expresión:
Ecuación
11:
Vm = (Lp -La) O
(b) Caso 2 Si se
cambian uno o más componentes durante la corrida de toma de muestra. En este
caso, reemplace Vm en la ecuación 9 con la expresión:
Ecuación
12
![]()
y substituya solamente por las fugas (Li o Lp)
que excedan a La.
12.4
Factores de conversión.
|
De |
a |
Multiplicar por |
|
scf |
m3 |
0.02832 |
|
g/ft3 |
gr/ft3 |
15.43 |
|
g/ft3 |
lb/ft3 |
2.205 * 10-3 |
|
g/ft3 |
g/m3 |
35.31 |
12.5 Variación
isocinética.
12.5.1
Cálculo a partir de los datos crudos:
Ecuación
13
![]()
donde:
K3
= 0,003454 mm Hg-m3/ml-°K.
12.5.2 Cálculo a partir
de valores intermedios:
Ecuación
15
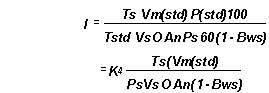
donde:
K4 = 4,320
12.6 Resultados
aceptables: Si 90% < I <110 por ciento los resultados son aceptables. Si
se encuentra un sesgo o desviación en el resultado por ejemplo I < 90%,
quiere decir que el resultado es menor al valor determinado y puede o no
aceptarse. Si el resultado presenta un sesgo o desviación I > 110%, quiere
decir que el valor es mayor al valor determinado y puede o no aceptarse el
resultado.
12.7 Para cálculo de
concentración de dioxinas y furanos
12.7.1 Factor de
Respuesta Relativa Promedio.
Ecuación
16
![]()
donde:
RRFi = Factor
de Respuesta Relativo del Compuesto “i”
n = No. Total de
Estándares de Calibración “j” Utilizados.
Acij = Corriente Iónica Integrada de los 2 Iones Característicos del
Compuesto “i” en el Estándar de Calibración “j”.
Acij* = Corriente
Iónica Integrada de los 2 Iones Característicos del Estándar Interno “i” en el
Estándar de Calibración “j”.
mci* = Masa
del Compuesto Etiquetado “i” en el Estándar de Calibración Inyectado al
Analizador, (pg).
mci = Masa
del Compuesto “i” en el Estándar de Calibración Inyectado al Analizador, (pg).
12.7.2. Masa
de PCDD’s y PCDF’s.
Ecuación 17
![]()
mi = Masa
del Compuesto “i” en la Muestra, (pg)
mi* = Masa
del Estándar Interno “i” Agregada a la Muestra, (pg)
RRFi = Factor
de Respuesta Relativo del Compuesto “i”.
Ai = Corriente
Iónica Integrada de los 2 Iones Característicos del Compuesto “i” en la
muestra.
Ai* = Corriente
Iónica Integrada de los 2 Iones Característicos del Estándar Interno “i” de la
muestra.
12.7.3. Masa
Total de PCDD’s y PCDF’s.
Ecuación 18
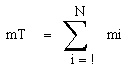
mT = Masa
Total de PCDD’s y PCDF’s en la Muestra, (pg)
mi = Masa
del Compuesto “i” en la Muestra, (pg)
N = No. Total de Compuestos.
NOTA: Si
algún PCDD o PCDF se encuentra por debajo del mínimo detectable, se deberá
considerar como cero para el cálculo del masa total de PCDD’s y PCDF’s.
12.7.4.
Cálculo de Concentración y Emisión con corrección a equivalentes tóxicos.
Siga los principios de cálculo de
concentración y emisión indicados en las secciones de PST y Metales, haciendo
la conversión de unidades apropiadas.
Corrección a Equivalentes Tóxicos de
Concentraciones de Dioxinas y Furanos.
Ci, EQT = Ci x Fi, EQT
Donde Fi es: Factor de equivalencia tóxica.
Concentración Equivalente Total y Factores de
Equivalencia Tóxica.
El procedimiento utilizado para el cálculo de
la Concentración Equivalente Tóxica Total fue basado en el siguiente documento:
"Exposure and Human Health Reassessment of
2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) and Related Compounds." U.S. Environmental Protection Agency's
(EPA) Part II Chapter 9: Toxic Equivalency Factors (TEF’s) for Dioxin and
Related Compounds” http://www.epa.gov/nceawww1/pdfs/dioxin/dioxreass.htm.
Los
factores de equivalencia tóxica al 2.3,7,8-Cl4-Dibenzo-p-Dioxina,
la cual posee un factor de toxicidad equivalente a 1.0, utilizados en este reporte, corresponden a la siguiente
referencia: U.S. Environmental Protection Agency's (EPA) TEF Values
http://www.epa.gov/nceawww1/dchem.htm
A
continuación se muestra una tabla indicando estos valores:
|
Compuesto |
Factor
de Equivalencia Tóxica |
|
Dioxinas 2,3,7,8-TCDD 1,2,3,7,8-PeCDD 1,2,3,4,7,8-HxCDD 1,2,3,6,7,8-HxCDD 1,2,3,7,8,9-HxCDD 1,2,3,4,6,7,8-HpCDD 1,2,3,4,6,7,8,9-OCDD |
1.0 0.5 0.1 0.1 0.1 0.01 0.001 |
|
Furanos 2,3,7,8-TCDF 1,2,3,7,8-PeCDF 2,3,4,7,8-PeCDF 1,2,3,4,7,8-HxCDF 1,2,3,6,7,8-HxCDF 1,2,3,7,8,9-HxCDF 2,3,4,6,7,8-HxCDF 1,2,3,4,6,7,8-HpCDF 1,2,3,4,7,8,9-HpCDF 1,2,3,4,6,7,8,9-OCDF |
0.1 0.05 0.5 0.1 0.1 0.1 0.1 0.01 0.01 0.001 |
Siga
los principios de cálculo indicados antes, haciendo las conversiones necesarias
para el manejo de las masas mT y ml en picogramos.
12.7.5 Mínimos de
Detección.
Cuando
el resultado del análisis de algún compuesto “i”, da por debajo del mínimo
detectable, se deberá reportar la concentración y emisión de dicho compuesto
como ‘menor a’ el resultado del mínimo detectable expresado como concentración
a condiciones de chimenea.
12.7.6 Criterios para
Promedios.
Dado
que generalmente un muestreo se compone de más de una muestra definitiva, es
necesario establecer los criterios a utilizar para el cálculo del promedio
global de concentración y emisión obtenidas en todos los resultados. Para lo
anterior: (a) el promedio de concentración y emisión de algún compuesto “i”
equivale al promedio aritmético de todos los definitivos ejecutados; (b) en
caso de que uno o más de los definitivos posean un resultado ‘menor a’
(equivalente al mínimo detectable del análisis, expresado a condiciones de
chimenea), y uno o más de los definitivos posean un resultado normal, para el
promedio global se deberán de considerar los resultados ‘menor a’ con magnitud
de cero, y; (c) en caso de que todos los definitivos posean un resultado ‘menor
a’, se deberá calcular su promedio con la magnitud indicada y expresar este
promedio como ‘menor a’.
12.8 La diferencia
porcentual relativa (DPR) se calcula como sigue:
Ecuación
19
DPR = S1 - S2/(S1 + S2)]*100
donde:
S1 y
S2 representan los resultados de la muestra y la muestra duplicada.
12.9 Reporte los
resultados en nanogramos por gramo con tres cifras significativas.
12.10
Cuando se analizan muestras adicionadas y duplicadas, todos los datos obtenidos
deben reportarse.
13.
Desempeño del Método
13.1
Límite de Detección: El mínimo detectable expresado en concentración a
condiciones del ducto en EQT, para los 17 congéneres en suma deberá ser menor o
igual al 10% del límite máximo permisible establecido en la Norma Oficial
correspondiente, en caso de no cumplir este criterio se deberán aplicar las
medidas correctivas necesarias tanto en el proceso analítico y de muestreo que
garanticen que se cumple esta disposición.
13.2
Límite Práctico de Cuantificación: Por determinar
13.3
Rango de Trabajo: Por determinar
13.4
Precisión Inicial del Método: Por determinar
13.5
Exactitud Inicial del Método: Por determinar
13.6
Precisión Continua del Método: Por determinar
13.7
Exactitud Continua del Método: Por determinar
14.
Prevención de la Contaminación
14.1 Los
solventes usados en este método plantean una pequeña amenaza al ambiente cuando
se manipulan adecuadamente. Las técnicas de evaporación de solventes utilizados
en este método son adecuadas para la recuperación de solventes y se recomienda
que el laboratorio recupere los solventes si esto es posible.
14.2 Los
estándares deben prepararse en volúmenes consistentes con el uso del laboratorio
para minimizar la disposición de estándares.
15.
Manejo de residuos
15.1 Es
la responsabilidad del laboratorio cumplir con todos los reglamentos federales,
estatales y locales referentes al manejo de residuos, particularmente las
reglas de identificación, almacenamiento y disposición de residuos peligrosos.
15.2 Las
muestras que contienen HCl a un pH < 2 son peligrosas y deben neutralizarse
antes de ser vertidas a un drenaje o debe manejarse como residuo peligroso.
15.3 Las
PCDDs/PCDFs se descomponen por arriba de 800ºC. Residuos de niveles bajos,
tales como papel absorbente, tejidos, restos de animales y guantes de plástico
pueden ser quemados en un incinerador apropiado. Grandes cantidades
(miligramos) deben empacarse con seguridad y disponerse a través de canales
comerciales o gubernamentales que son capaces de manejar residuos
extremadamente tóxicos.
15.4 Los
residuos líquidos y solubles deben disolverse en metanol o etanol e irradiarse
con luz ultravioleta con una longitud de onda corta de 290 nm por varios días.
Use una lámpara F40 BL o una equivalente. Analice los residuos líquidos y
disponga de las soluciones cuando los PCDDs/PCDFs no puedan ser detectados.
16. Bibliografía
16.1 Método
No. 8290, “Polychlorinated Dibenzodioxins (PCDDs and Dibenzofurans (PCDFs) by
High-resolution Gas Chromatography/High-resolution Mass Spectrometry”,
Environmental Protection Agency, November 1990
17.
Tablas y Figuras
Tabla 17.1
Compuestos que pueden determinarse por este método
|
Analito |
# CAS |
|
2,3,7,8-Tetraclorodibenzo-p-dioxina TCDD |
1740-01-6 |
|
1,2,3,7,8-Pentaclorodibenzo-p-dioxina PeCDD |
|
|
1,2,3,6,7,8-Hexaclorodibenzo-p-dioxina HxCDD |
|
|
1,2,3,4,7,8-Hexaclorodibenzo-p-dioxina HxCDD |
|
|
1,2,3,4,6,7,8-Heptaclorodibenzo-p-dioxina
HPPCDDs |
|
|
2,3,7,8-Tetraclorodibenzofurano TCDF |
|
|
1,2,3,7,8-Pentaclorodibenzofurano PeCDF |
|
|
2,3,4,7,8-Pentaclorodibenzofurano PeCDF |
|
|
1,2,3,6,7,8-Hexaclorodibenzofurano HxCDF |
|
|
1,2,3,7,8,9-Hexaclorodibenzofurano HxCDF |
|
|
1,2,3,4,7,8-Hexaclorodibenzofurano HxCDF |
|
|
2,3,4,6,7,8-Hexaclorodibenzofurano HxCDF |
|
|
1,2,3,4,6,7,8-Heptaclorodibenzofurano
HPPCDFs |
|
|
1,2,3,4,7,8,9-Heptaclorodibenzofurano
HPPCDFs |
|
Tabla 17.2 Tipos de matrices, tamaño de la
muestra y Límites de Calibración del Método basados en el 2,3,7,8-TCDD
|
|
Residuos acuosos |
Pulpa de Papel |
Cenizas |
Fondo fijo |
Sedimentos y
aceites |
|
LMC inferiora |
0.01 |
1.0 |
1.0 |
10 |
5.0 |
|
LMC superiora |
2 |
200 |
200 |
2,000 |
1,000 |
|
Peso (g) |
1,000 |
10 |
10 |
- |
2 |
|
Nivel de Adición IS (ppt) |
1 |
100 |
100 |
1,000 |
500 |
|
Volumen final de extracción (µL) |
10-50 |
10-50 |
50 |
50 |
50 |
|
|
|
|
|
|
|
a Para
los otros isómeros multiplique el valor por 1 para TCDD/PeCDD/PeCDF, por 2.5 para HxCDD/HxCDF/HPPCDDs/HPPCDFs y
por 5 para OCDD/OCDF.
17.3 Composición de las soluciones de
fortificación de la muestra y estándar de recuperacióna
|
Analito |
Concentración de
la solución de fortificación de la muestra (pg/µL; solvente: nonano) |
Concentración de
la solución estándar de recuperación (pg/µL; solvente: nonano) |
|
13C12-2,3,7,8-TCDD |
10 |
-- |
|
13C12-2,3,7,8-TCDF |
10 |
-- |
|
13C12-1,2,3,4-TCDD |
-- |
50 |
|
13C12-1,2,3,7,8-PeCDD |
10 |
-- |
|
13C12-1,2,3,7,8-PeCDD |
10 |
-- |
|
13C12-1,2,3,6,7,8-HxCDD |
25 |
-- |
|
13C12-1,2,3,4,7,8-HxCDF |
25 |
-- |
|
13C12-1,2,3,7,8,9-HxCDD |
-- |
50 |
|
13C12-1,2,3,4,6,7,8-HPPCDDs |
25 |
-- |
|
13C12-1,2,3,4,6,7,8-HPPCDFs |
25 |
-- |
|
13C12-OCDD |
50 |
-- |
|
|
|
|
a
Estas soluciones debe hacerse todos los días debido a la posibilidad de
pérdidas por absorción en el material de vidrio. Si estas soluciones serán
guardadas más de un día, entonces la solución de fortificación de muestras debe
incrementar la concentración 10 veces y la concentración de la solución
estándar de recuperación debe aumentar al doble. Ajuste los correspondientes
volúmenes de adición.
Tabla 17.4 Los 15
isómeros 2,3,7,8-sustituidos de las PCDDs y PCDFs
|
PPCDDS |
PPCDFS |
|
2,3,7,8-TCDD* |
2,3,7,8-TCDF* |
|
1,2,3,7,8-PeCDD* |
1,2,3,7,8-PeCDF* |
|
1,2,3,6,7,8-HxCDD* |
2,3,4,7,8-PeCDF |
|
1,2,3,4,7,8-HxCDD |
1,2,3,6,7,8-HxCDF |
|
1,2,3,7,8,9-HxCDD+ |
1,2,3,7,8,9-HxCDF |
|
1,2,3,4,6,7,8-HPPCDDs* |
1,2,3,4,7,8-HxCDF* |
|
|
2,3,4,6,7,8-HxCDF |
|
|
1,2,3,4,6,7,8-HPPCDFs* |
|
|
1,2,3,4,7,8,9-HPPCDFs |
|
|
|
* El análogo 13C-marcado se usa como un estándar interno.
+ El análogo 13C-marcado se usa como un estándar de
recuperación.
Tabla 17.5
Isómeros de Dioxinas y Furanos Clorados como una Función del Número de átomos
de cloro.
|
Número de átomos
de cloro |
Número de
isómeros de dioxinas |
Número de
isómeros 2,3,7,8 |
Número de
isómeros de furanos |
Número de
isómeros 2,3,7,8 |
|
1 |
2 |
--- |
4 |
--- |
|
2 |
10 |
--- |
16 |
--- |
|
3 |
14 |
--- |
28 |
--- |
|
4 |
22 |
1 |
38 |
1 |
|
5 |
14 |
1 |
28 |
2 |
|
6 |
10 |
3 |
16 |
4 |
|
7 |
2 |
1 |
4 |
2 |
|
8 |
1 |
1 |
1 |
1 |
|
|
|
|
|
|
|
Total |
75 |
7 |
135 |
10 |
|
|
|
|
|
|
17.6 Soluciones de
calibración de concentración de alta resolución.
|
Compuesto |
Concentración
(pg/µL en nonano) |
||||
|
|
5 |
4 |
3 |
2 |
1 |
|
Analitos no marcados |
|
|
|
|
|
|
2,3,7,8-TCDD |
200 |
50 |
10 |
2.5 |
|
|
2,3,7,8-TCDF |
200 |
50 |
10 |
2.5 |
|
|
1,2,3,7,8-PeCDD |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,7,8-PeCDF |
500 |
125 |
25 |
6.25 |
2.5 |
|
2,3,4,7,8-PeCDF |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,4,7,8-HxCDD |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,6,7,8-HxCDD |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,7,8,9-HxCDD |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,4,7,8-HxCDF |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,6,7,8-HxCDF |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,7,8,9-HxCDF |
500 |
125 |
25 |
6.25 |
2.5 |
|
2,3,4,6,7,8-HxCDF |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,4,6,7,8-HPPCDDs |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,4,6,7,8-HPPCDFs |
500 |
125 |
25 |
6.25 |
2.5 |
|
1,2,3,4,7,8,9-HPPCDFs |
500 |
125 |
25 |
6.25 |
2.5 |
|
OCDD |
1,000 |
250 |
50 |
12.5 |
5 |
|
OCDF |
1,000 |
250 |
50 |
12.5 |
5 |
|
|
|
|
|
|
|
|
Estándares Internos |
|
|
|
|
|
|
13C12-2,3,7,8-TCDD |
50 |
50 |
50 |
50 |
50 |
|
13C12-2,3,7,8-TCDF |
50 |
50 |
50 |
50 |
50 |
|
13C12-1,2,3,7,8-PeCDD |
50 |
50 |
50 |
50 |
50 |
|
13C12-1,2,3,7,8-PeCDF |
50 |
50 |
50 |
50 |
50 |
|
13C12-1,2,3,6,7,8-HxCDD |
125 |
125 |
125 |
125 |
125 |
|
13C12-1,2,3,4,7,8-HxCDF |
125 |
125 |
125 |
125 |
125 |
|
13C12-1,2,3,4,6,7,8-HPPCDDs |
125 |
125 |
125 |
125 |
125 |
|
13C12-1,2,3,4,6,7,8-HPPCDFs |
125 |
125 |
125 |
125 |
125 |
|
13C12-OCDD |
250 |
250 |
250 |
250 |
250 |
|
|
|
|
|
|
|
|
Estándares de Recuperación |
|
|
|
|
|
|
13C12-1,2,3,4-TCDDa |
50 |
50 |
50 |
50 |
50 |
|
13C12-1,2,3,7,8,9-HxCDDb |
125 |
125 |
125 |
125 |
125 |
|
|
|
|
|
|
|
a Utilizado para la
recuperación de estándares internos TCDD, TCDF, PeCDD y PeCDF.
b Usados para la
recuperación del estándar interno de HxCDD, HxCDF, HPPCDDs, HPPCDFs y OCDD.
Tabla 17.7 Iones
monitoreados para el Análisis de PCDDs/PCDFs por HRGC/HRMS
|
Descriptor |
Exactitud de
masasa |
ID del Ión |
Composición Elemental |
Analito |
|
1 |
303.9016 |
M |
C12H435Cl40 |
TCDF |
|
|
305.8987 |
M+2 |
C12H435C1337ClO |
TCDF |
|
|
315.9419 |
M |
13C12H435Cl40 |
TCDF (S) |
|
|
317.9389 |
M+2 |
13C12H35Cl337C10 |
TCDF (S) |
|
|
319.8965 |
M |
C12H4(35)C14O2 |
TCDD |
|
|
321.8936 |
M+2 |
C12H4(35)C13(37)Cl02 |
TCDD |
|
|
331.9368 |
M |
13C12H4(35)Cl4O2 |
TCDD (S) |
|
|
333.9338 |
M+2 |
13C12H4(35)Cl3(37)ClO2 |
TCDD (S) |
|
|
375.8364 |
M+2 |
C12H4(35)Cl5(37)ClO |
HxCDPE |
|
|
[354.9792] |
LOCK |
C9F13 |
PFK |
|
|
|
|
|
|
|
2 |
339.8597 |
M+2 |
C12H3(35)Cl4(37)ClO |
PeCDF |
|
|
341.8567 |
M+4 |
C12H3(35)Cl3(37)Cl20 |
PeCDF |
|
|
351.9000 |
M+2 |
13C12H3(35)Cl4(37)ClO |
PeCDF (S) |
|
|
353.8970 |
M+4 |
13C12H3(35)Cl3(37)Cl2O |
PeCDF (S) |
|
|
355.8546 |
M+2 |
C12H3(35)Cl4(37)C102 |
PeCDD |
|
|
357.8516 |
M+4 |
C12H3(35)C13(37)Cl202 |
PeCDD |
|
|
367.8949 |
M+2 |
13C12H3(35)C14(37)Cl02 |
PeCDD (S) |
|
|
369.8919 |
M+4 |
13C12H3(35)C13(37)Cl202 |
PeCDD (S) |
|
|
409.7974 |
M+2 |
C12H3(35)C16(37)ClO |
HpCDPE |
|
|
[354.9792] |
LOCK |
C9F13 |
PFK |
|
|
|
|
|
|
|
3 |
373.8208 |
M+2 |
C12H2(35)C15(37)ClO |
HxCDF |
|
|
375.8178 |
M+4 |
C12H2(35)C14(37)Cl2O |
HxCDF |
|
|
383.8639 |
M |
13C12H2(35)Cl60 |
HxCDF (S) |
|
|
385.8610 |
M+2 |
13C12H2(35)C15(37)ClO |
HxCDF (S) |
|
|
389.8156 |
M+2 |
C12H2(35)C15(37)Cl02 |
HxCDD |
|
|
391.8127 |
M+4 |
C12H2(35)C14(37)Cl202 |
HxCDD |
|
|
401.8559 |
M+2 |
13C12H2(35)C15(37)ClO2 |
HxCDD (S) |
|
|
403.8529 |
M+4 |
13C12H2(35)C14(37)Cl202 |
HxCDD (S) |
|
|
445.7555 |
M+4 |
C12H2(35)C16(37)Cl20 |
OCDPE |
|
|
[430.9728] |
LOCK |
C9F17 |
PFK |
|
|
|
|
|
|
|
4 |
407.7818 |
M+2 |
C12H(35)C16(37)ClO |
HPPCDFs |
|
|
409.7788 |
M+4 |
C12H(35)C15(37)Cl2O |
HPPCDFs |
|
|
417.8250 |
M |
13C12H(35)Cl70 |
HPPCDFs (S) |
|
|
419.8220 |
M+2 |
13C12H(35)C16(37)ClO |
HPPCDFs |
|
|
423.7767 |
M+2 |
C12H(35)C16(37)ClO2 |
HPPCDDs |
|
|
425.7737 |
M+4 |
C12H(35)C15(37)Cl202 |
HPPCDDs |
|
|
435.8169 |
M+2 |
13C12H(35)C16(37)ClO2 |
HPPCDDs (S) |
|
|
437.8140 |
M+4 |
13C12H(35)C17(37)Cl202 |
HPPCDDs (S) |
|
|
479.7165 |
M+4 |
C12H5C173Cl20 |
NCDPE |
|
|
[430.9728] |
LOCK |
C9F17 |
PFK |
|
|
|
|
|
|
|
5 |
441.7428 |
M+2 |
C12(35)C17(37)CLO |
OCDF |
|
|
443.7399 |
M+4 |
C12(35)C16(37)Cl20 |
OCDF |
|
|
457.7377 |
M+2 |
C12(35)C17(37)ClO2 |
OCDD |
|
|
459.7348 |
M+4 |
C12(35)C16(37)Cl202 |
OCDD |
|
|
469.7780 |
M+2 |
13C12(35)C17(37)ClO2 |
OCDD (S) |
|
|
471.7750 |
M+4 |
13C12(35)C16(37)Cl2O2 |
OCDD (S) |
|
|
513.6775 |
M+4 |
C12(35)C18(37)Cl2O |
DCDPE |
|
|
[442.9278] |
LOCK |
C10F17 |
PFK |
|
|
|
|
|
|
Las
siguientes masas nuclídicas fueron usadas:
H = 1.007825 O = 15.994915
C = 12.0000 (35)
Cl = 34.968853
13C = 13.003355 Cl = 36.965903
F = 18.9984
S = estándar
de recuperación/estándar interno
Tabla 17.8 Isómeros presente de PCDDs y PCDFs en
la solución de evaluación del desempeño y uso para definición de los intervalos
de los tiempos de retención del GC en una columna 60 m DB-5.
|
Número de átomos
de cloro |
Posición del
isómero de PPCDDS |
Posición del
isómero de PPCDFS |
||
|
|
Primer Eluado |
Ultimo Eluado |
Primer Eluado |
Ultimo Eluado |
|
4a |
1,2,6,8 |
1,2,8,9 |
1,3,6,8 |
1,2,8,9 |
|
5 |
1,2,4,6,8/ 1,2,4,7,9 |
1,2,3,8,9 |
1,3,4,6,8 |
1,2,3,8,9 |
|
6 |
1,2,4,6,7,9/ 1,2,4,6,8,9 |
1,2,3,4,6,7 |
1,2,3,4,6,8 |
1,2,3,4,8,9 |
|
7 |
1,2,3,4,6,7,9 |
1,2,3,4,6,7,8 |
1,2,3,4,6,7,8 |
1,2,3,4,7,8,9 |
|
8 |
|
1,2,3,4,6,7,8,9 |
|
1,2,3,4,6,7,8,9 |
|
|
|
|
|
|
a En adición a
estos dos isómeros TCDD, los isómeros 1,2,3,4-, 1,2,3,7-, 1,2,3,8-, 2,3,7,8- 13C12-2,3,7,8- y
1,2,3,9-TCDD debe presentarse también como una verificación de la resolución de
la columna.
Tabla 17.9 Proporción teórica de la abundancia
de los iones y sus límites de control para
PCDDs y PCDFs
|
Número de átomos
de cloro |
Tipo de ión |
Proporción
teórica |
Límites de
Control |
|
|
|
|
|
Inferior |
Superior |
|
4 |
M/M+2 |
0.77 |
0.65 |
0.89 |
|
5 |
M+2/M+4 |
1.55 |
1.32 |
1.78 |
|
6 |
M+2/M+2 |
1.24 |
1.05 |
1.43 |
|
6ª |
M/M+2 |
0.51 |
0.43 |
0.59 |
|
7b |
M/M+2 |
0.44 |
0.37 |
0.51 |
|
7 |
M+2/M+4 |
1.04 |
0.88 |
1.20 |
|
8 |
M+2 |
0.89 |
0.76 |
1.02 |
|
|
|
|
|
|
a Usado
sólo para 13C-HxCDF
(IS).
b Usado sólo para 13C-HPPCDFs (IS).
Tabla 17.10 Atribuciones de Factores de
Respuesta Relativo [RRF (número)].
|
Número |
Nombre del
congénere específico |
|
1 |
2,3,7,8-TCDD (y TPCDDs totales) |
|
2 |
2,3,7,8-TCDF (y TPCDFs totales) |
|
3 |
1,2,3,7,8-PeCDD (y PePCDDs totales) |
|
4 |
1,2,3,7,8-PeCDF |
|
5 |
2,3,4,7,8-PeCDF |
|
6 |
1,2,3,4,7,8-HxCDD |
|
7 |
1,2,3,6,7,8-HxCDD |
|
8 |
1,2,3,7,8,9-HxCDD |
|
9 |
1,2,3,4,7,8-HxCDF |
|
10 |
1,2,3,6,7,8-HxCDF |
|
11 |
1,2,3,7,8,9-HxCDF |
|
12 |
2,3,4,6,7,8-HxCDF |
|
13 |
1,2,3,4,6,7,8-HPPCDDs (y HPPCDDss totales) |
|
14 |
1,2,3,4,6,7,8-HPPCDFs |
|
15 |
1,2,3,4,7,8,9-HPPCDFs |
|
16 |
OCDD |
|
17 |
OCDF |
|
18 |
13C12-2,3,7,8-TCDD |
|
19 |
13C12-2,3,7,8-TCDF |
|
20 |
13C12-1,2,3,7,8-PeCDD |
|
21 |
13C12-1,2,3,7,8-PeCDF |
|
22 |
13C12-1,2,3,6,7,8-HxCDD |
|
23 |
13C12-1,2,3,4,7,8-HxCDF |
|
24 |
13C12-1,2,3,4,6,7,8-HPPCDDs |
|
25 |
13C12-1,2,3,4,6,7,8-HPPCDFs |
|
26 |
13C12-OCDD |
|
27 |
PeCDF’s totales |
|
28 |
HxCDF’s totales |
|
29 |
HxCDD’s totales |
|
30 |
HPCDFs totales |
|
|
|
Tabla 17.11 Factores de equivalencia de
toxicidad (TEFs) para el 2,3,7,8-TCDD para los dibenzodioxinas y dibenzofuranos
policlorados.
|
Número |
Compuestos |
TEF |
|
1 |
2,3,7,8-TCDD |
1.00 |
|
2 |
1,2,3,7,8-PeCDD |
0.50 |
|
3 |
1,2,3,6,7,8-HxCDD |
0.10 |
|
4 |
1,2,3,7,8,9-HxCDD |
0.10 |
|
5 |
1,2,3,4,7,8-HxCDD |
0.10 |
|
6 |
1,2,3,4,6,7,8-HPPCDDs |
0.01 |
|
7 |
1,2,3,4,6,7,8,9-OCDD |
0.001 |
|
|
|
|
|
8 |
2,3,7,8-TCDF |
0.1 |
|
9 |
1,2,3,7,8-PeCDF |
0.05 |
|
10 |
2,3,4,7,8-PeCDF |
0.5 |
|
11 |
1,2,3,6,7,8-HxCDF |
0.1 |
|
12 |
1,2,3,7,8,9-HxCDF |
0.1 |
|
13 |
1,2,3,4,7,8-HxCDF |
0.1 |
|
14 |
2,3,4,6,7,8-HxCDF |
0.1 |
|
15 |
1,2,3,4,6,7,8-HPPCDFs |
0.01 |
|
16 |
1,2,3,4,7,8,9-HPPCDFs |
0.01 |
|
17 |
1,2,3,4,6,7,8,9-OCDF |
0.001 |
|
|
|
|
Tabla 17.12 Tiempos de retención relativa de
los analitos de los atributos de referencia.
|
Analito |
RRF del analito
de referenciaa |
|
1,2,3,4,7,8-HxCDD |
13C12-1,2,3,6,7,8-HxCDD |
|
1,2,3,6,7,8-HxCDF |
13C12-1,2,3,4,7,8-HxCDF |
|
1,2,3,7,8,9-HxCDF |
13C12-1,2,3,4,7,8-HxCDF |
|
2,3,4,6,7,8-HxCDF |
13C12-1,2,3,4,7,8-HxCDF |
a Los tiempos de
retención del 2,3,4,7,8-PeCDF en la columna DB-5 se mide relativo al 13C12-1,2,3,7, 8-PeCDF
y el tiempo de retención del 1,2,3,4,7,8,9-HPPCDFs relativo al 13C12-1,2,3,4,6,7,8-HPPCDFs.
ANEXO 5B
METODO DE
ANALISIS PARA DETERMINAR POLICLORODIBENZO-p-DIOXINAS (PCDDs) Y POLICLORODIBENZOFURANOS (PCDFs) EN
EMISIONES A LA ATMOSFERA DE CHIMENEAS
POR CROMATOGRAFIA DE GASES ALTA RESOLUCION ACOPLADO A ESPECTROMETRIA DE
MASAS DE BAJA RESOLUCION HRGC/LRMS
Declaraciones
La mención de nombres de negocios o productos
comerciales no constituye un apoyo o recomendación para su uso.
Introducción
Este método está basado en su desempeño, lo
cual permite que a futuro no quede obsoleto, ya que podrá actualizarse
automáticamente siempre y cuando se aplique para la misma matriz y rango de
trabajo.
El principio de este método se basa en la
adición de estándares internos isotópicamente marcados a las muestras en
concentraciones conocidas, su extracción, fraccionamiento y limpieza utilizando
columnas cromatográficas secuencialmente y el análisis del extracto procesado
para las Policlorodibenzo-p-dioxinas y Policlorodibenzofuranos (PCDDs y PCDFs)
utilizando Cromatografía de Gases de Alta Resolución acoplada a Espectrometría
de Masas de Baja Resolución (HRGC/LRMS).
La base de este método está tomada del Método
428, “Determination of Polychlorinated Dibenzo-p-Dioxin (PCDD) and
Polychlorinated Dibenzofuran (PCDF) in Emissions from Stationary Sources”,
State of California, Air Resources Board, March 1988 y 23 A de la USEPA
“Determination of Polychlorinated Dibenzo-p-Dioxins and Polychlorinated
Dibenzofurans in Emissions from Stationary Sources”.
NOTA: Este
método está basado en su desempeño, se permite que el laboratorio omita
cualquier paso o modifique cualquier procedimiento, suponiendo que todos los
requerimientos de desempeño especificados se cumplan. Al laboratorio no se le
permite omitir cualquier punto de control de calidad, ni los parámetros que se
especifiquen como “NO MODIFICABLES”. Los términos “debe”, “puede” y “deberá”
son mencionados a través de los métodos y están destinados a ilustrar la importancia
de los procedimientos para producir datos verificables en los rangos de trabajo
del método. El término “debe” es usado para indicar que los desarrolladores de
este método encontraron ciertos procedimientos esenciales en muestras
analizadas exitosamente; de todas maneras, estos procedimientos pueden ser
modificados u omitidos si el laboratorio puede demostrar fehacientemente que la
calidad de los resultados no resulta afectada. Los requerimientos para
establecer la equivalencia del método se encuentran en la sección 9.0.
Parámetros
no modificables
Cromatografía de Gases con Columna Capilar y
Detector de Espectrometría de Masas
Contenido
1.
Aplicación y Alcances
2.
Resumen
3.
Definiciones
4.
Interferencias
5.
Seguridad
6.
Equipo y Materiales
7.
Reactivos y Patrones
8.
Recolección, Preservación y Almacenamiento de Muestras
9.
Control de Calidad
10.
Calibración
11.
Procedimiento
12.
Cálculos
13.
Desempeño del Método
14.
Prevención de la Contaminación
15.
Manejo de Residuos
16.
Referencias
17.
Tablas y Figuras
1.
Aplicación y Alcances
1.1
Propósito
Este método está diseñado para la medición de
las PCDDs/PCDFs en emisiones a la atmósfera de chimeneas de incineradores de
residuos o chimeneas de procesos industriales, está basado en el muestreo
isocinético de las corrientes de gases de la chimenea y la posterior
determinación por HRGC/LRMS.
1.2
Analitos
Este método es aplicable a la determinación
cualitativa y cuantitativa de los 17 isómeros 2,3,7,8 sustituidos de las
PCDDs/PCDFs y a la determinación cuantitativa en forma total por grado de
cloración de los demás isómeros.
1.3
Matrices
Este método es aplicable sólo a las emisiones
a la atmósfera de chimeneas ya sea de incineradores de residuos o de procesos
industriales.
1.4
Limitaciones
El límite de detección del método se da en el
rango de picogramos a nanogramos por m3, pero la sensibilidad dependerá finalmente
del volumen de muestra tomado y de las interferencias de matriz.
1.5
Restricciones
El uso de este método está restringido a
analistas instrumentales de amplia experiencia en el manejo de columnas
capilares, cromatógrafos de gases, espectrómetros de masas y en la
interpretación de espectros de masas por SCAN y por SIM. Cada analista debe
demostrar la habilidad para generar resultados aceptables con este método
usando los procedimientos establecidos en la Sección 9.
2.
Resumen
2.1 Las
PCDDs y PCDFs en forma de partículas y en forma gaseosa son muestreadas en la
chimenea isocinéticamente y colectadas en un filtro y en un cartucho de resina
XAD-2 previamente fortificado con estándares surogados marcados isotópicamente,
posteriormente son recuperadas de las diversas partes del tren de muestreo en
el campo y los enjuagues son transportados al laboratorio para su análisis.
Los enjuagues son extraídos y concentrados,
los extractos resultantes son fortificados con estándares internos marcados
isotópicamente y fraccionados en varias columnas cromatográficas que contienen
adsorbentes específicos para remover los compuestos interferentes más comunes,
los extractos limpios son concentrados a 50 L y se les agregan
estándares de recuperación marcados isotópicamente, posteriormente se inyectan
2L en el sistema
HRGC/LRMS para su identificación y cuantificación.
2.2
Varios criterios de optimización se especifican en el presente método los
cuales deben cubrirse para asegurar la calidad analítica de los resultados.
2.3 Este
método tiene el objetivo de determinar la concentración total de dioxinas y
furanos y sus isómeros. Esto significa que se determinarán desde las tetracloro
hasta las octaclorodibenzo-p-dioxinas y furanos. Este método se aplica a los 17
isómeros 2,3,7,8 sustituidos. De los 75 isómeros de Dioxinas y 135 isómeros de
Furanos, se encuentran en este método 22 TCDD, 38 TCDF, 14 PeCDD, 28 PeCDF, 10
HxCDD, y 16 HxCDF, 2 HpCDD, 4 HpCDF, 1 OCDD y 1 OCDF. De estos isómeros, los
que se presentan en la Tabla 17.1
representan los isómeros substituidos 2,3,7,8.
2.4 Los
límites de detección del método se muestran en la Tabla 17.2.
3.
Definiciones
Las definiciones presentadas en esta sección
son específicas para este método, pero han sido conformadas para que sean en lo
posible de uso común.
3.1
Blanco de calibración
Volumen de agua reactivo que se utiliza para
calibrar el instrumento. El blanco de calibración es un estándar cero.
3.2
Blanco de campo
Alícuota de agua reactivo que es colocada en
un envase para muestra en el laboratorio, empacada para el muestreo, y tratada
como una muestra en todos los aspectos, incluyendo el contacto con los equipos
de campo y expuesta a las condiciones del sitio de muestreo, almacenaje,
preservación y todos los procedimientos analíticos, los cuales pueden incluir
filtración. El propósito del blanco de campo es determinar si el ambiente del
campo o durante el transporte y almacenamiento de la muestra existe
contaminación por difusión de compuestos volátiles en la muestra.
3.3
Blanco de reactivos
Es una matriz libre de analitos a la cual se
le agregan todos los reactivos en los mismos volúmenes o proporciones usados en
el procesamiento de la muestra. El blanco de reactivos debe llevarse a través
de la preparación de la muestra y el procedimiento analítico. El blanco de
reactivos se usa para documentar la contaminación resultante del proceso
analítico.
3.4
Congénere
Se refiere a cualquiera de los compuestos
particulares de una misma familia química, p. e. existen 75 congéneres de las
PCDDs.
3.5
Desviación estándar(s)
Cuando se utiliza este estadístico en el
presente método, se refiere a la desviación estándar de la muestra, calculada a
partir de n-1 y no a la de la población () la cual se
calcula a partir de n.
3.6
Estándar interno
Solución preparada de un estándar diluido y/o
una solución patrón agregada a cada muestra, blanco muestra de control de
calidad, en la misma concentración, se agrega antes de iniciar el proceso de
extracción y se utiliza para medir la concentración de los analitos y de los
estándares surrogados.
3.7
Estándar subrogado
Solución preparada de un estándar diluido de
compuestos marcados isotópicamente y que se agrega a la resina XAD-2 antes de
armar el tren de muestreo, se utiliza para medir la eficiencia del muestreo y
de la recuperación de la muestra en campo.
3.8
Estándar de recuperación
Solución preparada de un estándar diluido de
compuestos marcados isotópicamente, se agrega antes de la inyección del
extracto final de las muestras, blancos y muestras de control de calidad y se
utiliza para medir la eficiencia en el proceso de extracción y limpieza del
extracto.
3.9
Homólogo
Grupo de sustancias estructuralmente
relacionadas (isómeros) con la misma fórmula molecular, p.e. existen 8
homólogos de las dioxinas, de la monoclorodibenzo-p-dioxina a la octaclorodibenzo-p-dioxina.
3.10
Isómero específico
Cualquiera de los isómeros en el que se indica
la posición exacta donde los átomos de cloro están situados, p.e. 2,3,7,8-TCDD
es uno de los 22 posibles isómeros de la TCDD.
3.11
Límite de Detección del Método (LDM)
Concentración mínima de un analito que puede
identificarse, medirse y reportarse con una confianza de 99% cuando la
concentración del analito es mayor a cero.
3.12
Límite práctico de cuantificación (LPC)
Concentración mínima del analito que puede
determinarse con un nivel de confianza predeterminado en condiciones rutinarias
de operación. Este límite puede establecerse entre 3 a 5 veces el LDM.
3.13
Muestra de Control de Calidad (MCC)
Muestra sintética que contiene todos o un
subgrupo de los analitos del método a una concentración conocida. La MCC se
obtiene de una externamente al laboratorio o es preparada de una fuente
diferente de los estándares de calibración. Se usa para revisar el desempeño
del laboratorio con materiales de prueba preparados externamente a los procesos
normales de preparación.
3.14
Rango de Trabajo
Rango de la concentración sobre el cual la
respuesta del instrumento para el analito es proporcional.
4.
Interferencias
4.1 Las
interferencias del método pueden ser causadas por contaminación de reactivos,
disolventes, material de vidrio y otros materiales que intervienen en el
procesamiento de la muestra. La contaminación puede causar un elevado nivel de
la señal de fondo del sistema.
Todo el material que interviene en el procesamiento
de la muestra debe demostrarse que está libre de interferencias bajo las
condiciones de análisis por medio del análisis de blancos de reactivos.
4.2 El
uso de reactivos de alta pureza ayuda a disminuir el problema de
interferencias.
4.3 Las
interferencias de matriz son causadas por contaminantes que son coextraídos de
la muestra. La variedad de interferencias de matriz es muy amplia ya que varía
dependiendo de la fuente que fue muestreada. Los PCDDs y PCDFs están
generalmente asociadas con otros compuestos clorados interferentes en
concentraciones mayores en hasta varios órdenes de magnitud que los compuestos
de interés. Los procedimientos de limpieza de la sección 11 se utilizan para
reducir muchas de estas interferencias, pero algunas muestras pueden requerir
limpiezas extraordinarias adicionales o una instrumentación con mayor poder de
resolución para obtener resultados confiables, la espectrometría de masas de
alta resolución puede usarse para evitar dar falsos positivos y mejorar los
límites de detección.
4.4 Se
deben emplear las siguientes columnas cromatográficas de alta resolución, DB-5
de 60 m y SP-2331 son las recomendadas para analizar las PCDDs/PCDFs. Ninguna
de las dos columnas resuelven todos los isómeros de las dioxinas y furanos.
Ambas columnas son requeridas para cuantificar todos los isómeros 2,3,7,8
sustituidos. La obtención de resultados positivos debe confirmarse utilizando
la otra columna.
5.
Seguridad
5.1 La
carcinogenicidad de todos los reactivos no ha sido determinada con precisión;
de todas maneras cada sustancia química debe tratarse como potencial peligro a
la salud. La exposición a estas sustancias químicas debe reducirse al menor
nivel posible. Se sugiere que el laboratorio realice monitoreos de higiene
ocupacional de cada reactivo a los que pueda estar expuesto el analista y que
dichos resultados estén disponibles para los analistas.
5.1.1 Se
ha encontrado que el isómero 2,3,7,8-TCDD es un carcinógeno y teratógeno en
estudios con animales de laboratorio. Es soluble en agua aproximadamente a 200
ppt y en disolventes orgánicos en un 0.14%.
Con base en la disponibilidad de información toxicológica y las propiedades
físicas del 2,3,7,8-TCDD, todos los PCDDs/PCDFs deben manejarse sólo por
personal altamente entrenado y familiarizado con el manejo y procedimientos de
precaución y con conocimientos de los riesgos asociados.
5.1.2 Se
recomienda que el laboratorio maneje soluciones estándares diluidas de los
analitos de este método. Sin embargo, si se preparan soluciones primarias,
deben prepararse en una campana y debe utilizarse una mascarilla con filtro
para gases tóxicos.
5.2 Este
método puede no mencionar todas las precauciones de seguridad asociadas con su
uso. El laboratorio es responsable de mantener un ambiente de trabajo seguro y
un archivo de las normas de seguridad respecto a la exposición y manejo seguro
de las sustancias químicas especificadas en este método. Debe tenerse un
archivo de referencias de las hojas de información de seguridad el cual debe
estar disponible a todo el personal involucrado en estos análisis.
5.3 Los PCDDs/PCDFs y
muestras que se sospecha contienen estos compuestos deben manejarse usando
esencialmente las mismas técnicas empleadas en el manejo de material
radioactivo o infeccioso. Se requiere de un laboratorio bien ventilado y de
acceso controlado. Los PCDDs/PCDFs son extremadamente tóxicos para animales de
laboratorio. Cada laboratorio debe desarrollar un programa estricto de
seguridad para el manejo de estos compuestos.
5.3.1 Instalaciones
Deben
contar con un sistema de extracción de aire forzado separado de los demás del
laboratorio para evitar la contaminación cruzada entre diferentes áreas del
laboratorio.
5.3.2 Equipo de
protección
Deben
usarse guantes de plástico desechables, bata de laboratorio o delantal, lentes
de seguridad o máscara, y una caja de guantes o campana de extracción adecuada
para trabajo radiactivo. Durante las operaciones analíticas que pueden producir
aerosoles ascendentes o polvos, el personal debe usar equipos de respiración
con filtros de carbón activado. El equipo de protección de los ojos
(preferiblemente cubriendo la cara completa) debe usarse mientras se está
trabajando con muestras o estándares analíticos puros. Se deben utilizar
guantes de látex para reducir la exposición de las manos. Cuando se sospecha o
se conoce que las muestras manejadas contienen altas concentraciones de
PCDDs/PCDFs, un juego adicional de guantes puede también utilizarse debajo de
los guantes de látex.
5.3.3 Entrenamiento
Los
trabajadores deben entrenarse en el método apropiado para remover los guantes y
ropa contaminada sin contacto con las superficies exteriores.
5.3.4 Higiene personal
Las
manos y los antebrazos deben lavarse extensamente después de cada manipulación
y antes de cada alimento.
5.3.5 Confinamiento
Areas
de trabajo aisladas cerradas con señalamiento, material de vidrio separado y
marcado y papel plástico absorbente en las mesas de trabajo ayudan en el
confinamiento de contaminación.
5.3.6 Efluentes de
Vapores
Los
efluentes del cromatógrafo de gases a través de la válvula “split” y de la
bomba de vacío del espectrómetro de masas deben pasar a través de una columna
de carbón activado o ser burbujeado a través de una trampa que contenga aceite
o alcoholes de alto punto de ebullición para condensar los vapores de
PCDDs/PCDFs.
5.3.7 Manejo de
residuos
Una
buena técnica incluye la disminución de residuos contaminados. Se deben
utilizar bolsas de plástico para los residuos sólidos. El personal de limpieza
debe estar entrenado en el manejo seguro de los residuos.
5.3.8 Descontaminación
5.3.8.1 Descontaminación
del personal
Use
cualquier jabón suave con abundante acción de restregado.
5.3.8.2 Materiales de
vidrio, herramientas y superficies
El
cloroetano es el disolvente menos tóxico que ha mostrado ser efectivo. Una
limpieza satisfactoria puede completarse con un enjuague con cloroetano,
después lavar con detergente y agua. Si el material de vidrio se enjuaga
primero con disolvente, entonces el lavado con agua puede desecharse por el
desagüe. Dado el alto costo de la disposición, es prudente la disminución de
residuos de disolventes.
5.3.9 Lavandería
La
ropa que se conoce está contaminada, debe colectarse en bolsas de plástico. Las
personas que conduzcan las bolsas y laven la ropa deben ser avisadas del
peligro y entrenadas en el manejo apropiado. La ropa puede ponerse en una
lavadora, sin contacto, si la persona conoce de los problemas potenciales. La
lavadora debe trabajar un ciclo sin ropa antes de ser utilizada nuevamente.
5.3.10 Prueba de
limpieza
Un
método exitoso para determinar la limpieza de las superficies de trabajo y los
equipos es limpiar las superficies con un pedazo de papel filtro. La extracción
y análisis por GC con un detector de captura de electrones (ECD) pueden
permitir un límite de detección de 0.1
g; el análisis
usado en este método puede permitir un límite de detección aún más bajo.
Concentraciones menores de 0.1 µg
por papel indican una técnica de limpieza adecuada; cualquier otra
concentración más alta indica la necesidad de una limpieza mayor. Más de 10 µg
en el filtro constituye un peligro agudo y requiere una limpieza expedita de
los equipos o espacios de trabajo, e indica que han sido empleadas prácticas de
trabajo no aceptables.
6.
Equipo y materiales
La mención de marcas, modelos y proveedores de
equipos y materiales en este método se citan debido a que fueron los utilizados
para desarrollarlo y solamente tienen propósitos ilustrativos. Su mención no
implica ninguna aprobación oficial. Puede obtenerse un desempeño equivalente
usando otros equipos y materiales que no hayan sido especificados en este
método, pero la demostración del desempeño equivalente de otros equipos y
materiales es responsabilidad del laboratorio que utilice este método.
Sólo se mencionan los equipos y materiales que
son relevantes en este método analítico.
6.1 Tren
de muestreo
El tren consta de una boquilla, sonda,
calentador del filtro de partículas, condensador y módulo de resina absorbente
seguidos de 3 impactores y un cartucho de sílica gel desecante. Un ciclón puede
utilizarse en la caja caliente del filtro para usarse en chimeneas que emiten
una gran cantidad de partículas.
6.1.1
Boquilla
Una boquilla de acero inoxidable niquelada,
cuarzo o de vidrio de borosilicato. El ángulo de la punta debe ser de 30° y
debe cuidarse que mantenga el diámetro interno constante. Debe tenerse un rango
variado de tamaños de boquillas para muestreos isocinéticos por ejemplo de 0.32 a 1.27 cm o aun diámetros mayores si se requieren muestrear en
impactores y trenes de gran volumen. Los incrementos del diámetro de la
boquilla deben ser de 0.16 cm. Todas
las boquillas deben calibrarse de acuerdo a la Sección 10.1.1.
6.1.2
Sonda
La sonda debe ser de acero inoxidable
niquelado, teflón, vidrio de borosilicato o cuarzo. La superficie de la sonda
debe ser de un material inerte para las dioxinas y furanos y demás gases de la
chimenea. El material de la sonda debe ser inerte desde la boquilla hasta la
conexión con el filtro. Puede contar con una chaqueta para controlar la
temperatura y dar protección a la cubierta de la sonda. El material de la
cubierta de la sonda, debe ser de tal forma que permita conectarse y esté libre
de fugas, debe soportar vacío y permanecer sellado sin necesidad de grasa.
6.1.3
Línea de transferencia de la muestra
La línea de transferencia de la muestra debe
ser de teflón (1/4 in. OD * 1/32 in) sin uniones ni conexiones que permitan
fuga alguna y debe permanecer sellada aun con vacío y sin uso de grasa.
Asimismo, la línea debe ser lo más corta posible.
6.1.4
Portafiltro
El porta filtro debe ser de borosilicato y de
vidrio fritado, deberá sellar perfectamente vidrio a vidrio o con un anillo de
teflón sin requerir grasa. El soporte deberá estar inmediatamente después de la
sonda o ciclón dependiendo de la configuración usada.
6.1.5
Separador previo
Se puede usar un ciclón para remover las
partículas mayores antes de que el gas sea filtrado. El material debe ser
vidrio de borosilicato o cuarzo.
6.1.6
Condensador
El condensador debe ser de vidrio de
borosilicato y debe permitir el enfriamiento de los gases a menos de 20°C antes
de entrar al módulo de la resina.
6.1.7
Módulo de la resina
El módulo de la resina XAD-2 debe construirse
en vidrio y con conexiones que permitan un sello total aun aplicando vacío y
sin requerir grasa de silicona. Muchas trampas de resina verticales están
precedidas del condensador de serpentín. También orientado verticalmente, con
circulación de agua fría. El gas debe enfriarse a 20°C antes de entrar al
módulo que contiene la resina. La temperatura de los gases debe monitorearse
por un termopar colocado a la entrada de la resina. La resina debe estar
firmemente empacada para evitar la formación de canales durante la toma de
muestra. El módulo con la resina debe permanecer vertical durante todo el
muestreo.
6.1.8 Tren de
impactores.
Tres
o más impactores se deben conectar en serie con conexiones a prueba de fuga sin
el uso de grasa de silicona. Todos los impactores deben ser similares al diseño
de Greenburg-Smith modificados por el reemplazo de la punta con un tubo de 1.3 cm de diámetro interno que llegue a
1.3 cm del fondo del matraz.
El
primer impactor, deberá conectarse a la punta del módulo absorbente y éste debe
modificarse con una espiga corta para que la muestra de gas no burbujee en los
condensados colectados, este impactor debe estar vacío.
Debe
usarse un segundo impactor grande cuando se tienen contenidos de humedad altos
en el gas muestreado, ya que en este impactor se colectan los condensados que
pasan a través de la resina para su posterior análisis. El segundo impactor
inicialmente contiene agua o alternativamente 100 mL de etilenglicol el cual se
utiliza para retener dioxinas y furanos que no fueron retenidos en la resina.
El
tercer impactor debe estar vacío.
6.1.9 Cartucho de
sílica gel.
Debe
agregarse de 200 a 300 g de sílica gel para absorber la humedad y que no pase a
la bomba.
6.1.10 Tubo Pitot.
El
tubo Pitot tipo “S”, se coloca junto a la extensión de la sonda para que
permita monitorear constantemente la velocidad de los gases de la chimenea.
6.1.11 Medidor de
presión diferencial.
Dos
manómetros inclinados se utilizan, un manómetro debe usarse para la lectura de
la velocidad del frente (P) y el otro para
la lectura de la presión diferencial del orificio.
6.1.12 Sistema de
Medición.
Medidor
de vacío, bomba libre de fugas, termómetro con una exactitud de +- 3.0°C, medidor de gas seco con
variaciones máximas de 2% y demás equipo relacionado.
El
equipo normalmente utilizado es el recomendado para la determinación de
partículas en chimeneas con las modificaciones en la sonda, filtro y vidriería.
6.1.13 Barómetro.
Un
barómetro de mercurio u otro tipo que sea capaz de medir una presión
atmosférica con una precisión de 2.5
mm de Hg.
6.1.14 Sistema de
Calentamiento del Filtro.
El
sistema de calentamiento del filtro debe ser capaz de mantener el filtro
durante el muestreo a 120°C ± 14°C. El medidor de temperatura debe ser capaz de
medir con una exactitud de +- 3°C e instalado de tal manera que pueda
verificarse la temperatura durante todo el muestreo.
6.1.15 Filtros
Los
filtros utilizados en el muestreo deben ser de fibra de vidrio sin aglutinantes
orgánicos y deben tener al menos 99.5%
de eficiencia (0.05% de penetración)
en partículas de 0.3 µ de humo de
di-Octil ftalato.
Los
datos de control del fabricante, en este caso son suficientes. Antes de cada
muestreo cada lote de filtros debe ser sometido a limpieza y a control de
calidad y verificación de contaminación para demostrar que está limpio y que no
contiene nada que pueda interferir con el análisis.
La
limpieza de los filtros consiste en su extracción en Soxhlet en lotes menores a
50 filtros con los disolventes que se emplearán en el campo. Como aseguramiento
de calidad estos mismos disolventes deben concentrarse en la misma proporción
que el blanco y debe someterse a los mismos procedimientos de limpieza que la
muestra. La señal de fondo o valor del blanco observado debe convertirse a un
valor por filtro y debe corregirse por una diferencia en el factor de
concentración entre la muestra de chequeo (CFqc) y el análisis de la muestra
actual (CFs).
![]()
donde:
![]()
Los criterios cuantitativos para aceptar la
calidad de un filtro dependerán del límite de detección y los criterios
establecidos para la muestra y el programa analítico. Los filtros que den una
señal de fondo o señal del blanco por filtro mayor o igual al límite de
detección de la señal del analito deben rechazarse.
NOTA: Los
criterios de aceptación para la limpieza del filtro dependen no solamente del
límite de detección del método, sino también del volumen muestreado.
Si los filtros no pasan CC deben volver a
extraerse y el disolvente debe reanalizarse hasta que la señal de fondo sea
aceptablemente baja.
6.2
Recuperación de Muestra.
6.2.1
Cepillo para la Boquilla.
El cepillo para la boquilla debe ser de
material inerte con mango de acero inoxidable de tamaño adecuado especialmente
diseñado para las boquillas.
6.2.2
Botellas de Lavado.
Se recomiendan botellas de 500 mL de Nalgene.
6.2.3
Contenedores de vidrio para la muestra.
Los frascos de muestra deben ser de vidrio
ámbar de 500 o 1,000 mL con contratapa de teflón.
6.2.4
Contenedor para Guardar el Filtro.
Un frasco de vidrio prelavado con hexano y
tapa con contratapa de teflón.
6.2.5
Probeta de Vidrio o Balanza.
Para medir el agua condensada utilice una
probeta graduada de 500 mL con divisiones no mayor a 2 mL o utilice una balanza
capaz de pesar con una variación de 0.5
g.
6.2.6
Contenedor para Sílica Gel.
Para almacenar la sílica gel se requiere un
frasco de vidrio que al cerrarlo quede sellado.
6.3
Análisis.
El material de vidrio utilizado en el presente
procedimiento (incluido el aparato de Soxhlet y Botellas) debe lavarse tan
rápido como sea posible después de utilizarse. Deben lavarse con el último
disolvente utilizado. Después de lavar el material con disolvente debe lavarse
con agua caliente y detergente, enjuagarlo con agua desionizada, acetona,
tolueno y cloruro de metileno y secarlo a 120°C antes de ser nuevamente
utilizado.
6.3.1
Botella para la Muestra.
La botella debe ser vidrio ámbar de 125 a 250
mL con contratapa de teflón.
6.3.2
Kuderna Danish:
El tubo de Kuderna Danish debe estar graduado
ya que el volumen empleado en la prueba es importante. Así mismo debe tener
tapón para evitar las pérdidas por evaporación del extracto.
6.3.3
Matraz Concentrador de Kuderna Danish de 500 mL-Este matraz deberá contener
sujetador de resortes.
6.3.4
Columna Snyder-Columna Snyder de tres bolas.
6.3.5
Columna Snyder Micro de tres bolas.
6.3.6
Viales de 2 mL-Con inserto (para hacerlo microvial).
6.3.7
Soxhlet-Equipo Soxhlet con matraz de 1 l y mantilla de calentamiento; condensador
Allihn.
6.3.8
Rotavapor.
6.3.9
Turbovap-Evaporador con corriente de nitrógeno (N-Evap de Organomation
Associates, Turbovap de Zymark o equivalente).
6.3.10
Balanza Analítica con capacidad de pesar con exactitud de 0.0001 g.
6.3.11
Pipetas Pasteur de 5 ¾” por 7.0 mm.
6.3.12
Cromatógrafo de gases (HRGC).
Cromatógrafo de Gases con inyector capilar,
sistema Split-Splitless, marca Hewlett-Packard, modelo 5890 II o equivalente.
6.3.13
Columnas de Alta Resolución:
6.3.13.1
Columnas de sílica fundida de 60 m de longitud por 0.32 mm DI recubierta de DB-5 o equivalente. Esta columna debe
resolver cada uno de los homólogos de las PCDDs y PCDFs.
6.3.13.2 Una
columna capilar de sílica fundida de SP-2331 de 60 m de longitud, 0.32 mm de DI y 0.25 micras de espesor de película.
6.3.14
Espectrómetro de Masas (LRMS)
Espectrómetro de Masas de Baja Resolución
acoplado con una interfase directa al HRGC marca Hewlett-Packard modelo 5972
con sistema de adquisición de datos o equivalente.
7.
Reactivos y patrones
Los reactivos que requiere el método deben ser
tipo ACS grado reactivo o pesticida, a menos que otra cosa se indique.
Agua-El agua de referencia debe entenderse
como agua grado reactivo tipo I ASTM.
7.1
Muestreo
7.1.1
Resina XAD-2
El procedimiento
de limpieza de la resina se lleva a cabo en un Soxhlet gigante el cual va a
contener suficiente resina XAD-2.
Para varias trampas de muestreo. Los cartuchos deben ser de fibra de vidrio de
55 o 90 mm de diámetro interno por 150 mm de longitud y deberán contar con un
vidrio fritado en la parte alta, el cual debe estar empotrado 10 mm abajo de la
parte superior del cartucho para facilitar el drenaje. La resina debe retenerse
cuidadosamente en la copa del extractor con lana de vidrio, la limpieza de la
resina deberá realizarse en el siguiente orden.
|
DISOLVENTE |
PROCEDIMIENTO |
|
Agua |
1l, un ciclo y tirar el |
|
H2O |
|
|
Agua |
Extraer 8 h |
|
Alcohol metílico |
Extraer 22 h |
|
Cloruro de metileno |
Extraer 22 h |
|
Hexano |
Extraer 22 h |
Secar la resina XAD-2 por una de las
siguientes técnicas:
7.1.1.1
Después de evaluar muchas técnicas para remover el disolvente residual se
encontró que un lecho fluidizado es el más rápido.
7.1.1.2
Coloque la resina en una columna de 10 cm de diámetro interno y 60 cm de
longitud en la cual se colocan 500 g de resina y por la parte baja de la
columna se hace pasar un flujo de nitrógeno de alta pureza filtrado para
retener compuestos orgánicos, el cual mediante un serpentín se calienta máximo
a 40°C durante toda la noche a un flujo que fluidice la resina sin que se
pierda por el extremo superior.
7.1.1.3 El
almacenamiento de la resina limpia, seca y probada como se indica en la sección
7.1.1.4 puede realizarse durante
máximo 2 semanas en metanol grado pesticida. Cuando se requiera utilizar
nuevamente la resina durante el periodo de esas dos semanas se prepara la
resina drenando el metanol y enjuagando con cloruro de metileno y secando como
se indica en el punto 7.1.1.1.
PRECAUCION: La resina debe utilizarse en las
siguientes 24 h después de limpiarse. Sólo podrá guardarse por dos semanas en
las condiciones indicadas.
7.1.1.4 El
método para verificar la contaminación de la resina XAD-2 debe realizarse para
confirmar que está libre de contaminantes que interfieran con el análisis o
resten eficiencia de absorción. Debe hacerse la verificación de la resina
tomando una porción similar a la que se utilizará en el campo (50 g) y se
extraerá de la misma manera que se trata una muestra real y se analiza de igual
forma. El nivel de ruido o contaminantes que se acepten dependerá del nivel o
límite de detección requerido.
La resina que dé una señal de fondo igual o
mayor que el límite de detección del analito de interés deberá purificarse. El
criterio de aceptación de limpieza de la resina depende del límite de detección
inherente y del volumen de muestra que se tome en campo.
7.1.2
Sílica Gel-Utilice sílica gel con indicador de 6 a 16 mallas. La sílica gel
debe secarse previamente a su uso durante 2 h a 175°C.
7.1.3 Agua
Desionizada-El agua destilada y desionizada deberá guardarse en recipientes de
vidrio previamente enjuagado con hexano grado pesticida y con tapa de teflón.
7.1.4
Hielo en Trozos-Ponga hielo en trozos en el baño alrededor de los impactores.
7.1.5 Lana
de vidrio-Lana de vidrio lavada mediante 3 inmersiones en hexano y secada a
110°C. El almacenamiento de la lana de vidrio limpia y seca deberá ser en
frasco de vidrio lavado con hexano y con contratapa de teflón.
7.2
Recuperación de la Muestra.
7.2.1
Agua.
7.2.2
Acetona grado pesticida.
7.2.3 Hexano
grado pesticida.
7.2.4
Ciclohexano grado pesticida.
7.2.5
Tolueno grado pesticida.
7.2.6
Disulfuro de carbono grado pesticida.
7.2.7
Cloruro de metileno grado pesticida.
7.3
Análisis.
7.3.1
Agua.
7.3.2
Hexano grado pesticida.
7.3.3
Benceno grado pesticida.
7.3.4
Tolueno grado pesticida.
7.3.5
Tetradecano grado pesticida.
7.3.6
Alcohol metílico grado pesticida.
7.3.7
Cloruro de metileno grado pesticida.
7.3.8
Acido Sulfúrico H2SO4 gravedad
específica 1.84.
7.3.9
Sulfato de sodio secado a 400°C por 4 h. Lavado con cloruro de metileno y
secado nuevamente a 400°C por 2 h. Guárdelo en vidrio enjuagado con cloruro de
metileno y con tapa de teflón.
7.3.10
Sílica Gel para cromatografía en columna tipo 60 (o equivalente), marca EM
reactivo, 100/120 mallas lavado en Soxhlet con cloruro de metileno y activada a
120°C durante 12 h. Guárdela en un frasco de vidrio lavado con cloruro de
metileno y con tapa de teflón.
7.3.11
Sílica Gel Impregnada con Hidróxido de sodio-Mezcle 39 g de NaOH 1N con 100 g de
Sílica Gel y dispérsela con agitación hasta obtener una mezcla uniforme.
Guárdelo en un frasco de vidrio lavado con cloruro de metileno y con tapa de
teflón.
7.3.12
Sílica Gel impregnada con ácido sulfúrico. Ponga en un matraz con tapa tres
partes de Sílica Gel con dos partes de ácido sulfúrico concentrado y agite
hasta tener una mezcla uniforme. Guárdelo en un frasco de vidrio lavado con
cloruro de metileno y con tapa de teflón.
7.3.13
Carbopack C, 80/100 mallas, No. catálogo de Supelco 1-0258 o equivalente.
7.3.14
Celite 545, 80/100 Mallas-Celite 545 no lavada con ácido, Fisher C-212 o
equivalente.
7.3.15
Mezcla Carbopack/Celite, mezclar 16.4
g de Celite 545 con 3.6 g de
Carbopack C en un vialde 40 mL, activar por 6 h a 130°C y guardar en el
desecador.
NOTA: Si
el contenido de carbón de esta mezcla es mayor a 20% habrá baja recuperación de
analitos presentes en bajas concentraciones, por tal razón cada lote de
carbopack/celite deberá revisarse con el estándar de calibración de menor
concentración y deberá recuperarse al menos 50% de los analitos.
7.3.16
Alúmina ácida-Bio-Rad AG-4 No. Cat. 132-1240 o equivalente, lavada con cloruro
de metileno en un Soxhlet, secada y activada a 190°C durante 24 h.
NOTA: La
calidad óptima de la alúmina varía para cada fabricante y forma de
almacenamiento. Debe verificarse cada lote de alúmina para asegurarse que se
recuperan satisfactoriamente las PCDD’s y PCDF’s. Para esta prueba debe
utilizarse el nivel de calibración más bajo de chequeo.
7.3.17
Nitrógeno-Obtenido de un cilindro de nitrógeno cromatográfico o UAP.
7.3.18 Helio-UAP o
cromatográfico.
7.3.19 Soluciones
Estándar-Pueden emplearse estándares de soluciones comerciales: KOR isótopos
Fifty Six Rogers Street, Cambridge. MA. 02139 y Cambridge Isotope Laboratories
Inc., 141 Magazine Street, Cambridge MA. 02139 o equivalentes.
7.3.19.1 Solución Estándar
Patrón-Las soluciones comerciales vienen listas para su uso o tienen
instrucciones específicas para su dilución.
7.3.20 Estándares de
Calibración-Los estándares de calibración también vienen listos para su uso y
deben cumplir con las concentraciones de la Tabla 17.3.
7.3.21 Estándares
Internos (IS)-Idem 7.3.20.
7.3.22 Estándares de
Recuperación-Idem 7.3.20.
7.3.23 Solución de
evaluación de columna-La mezcla para la evaluación de la columna contiene los
isómeros listados abajo. Esta mezcla de isómeros es usada para definir los
tiempos de retención y las ventanas de tiempo de cada uno de los homólogos de
las dioxinas y los furanos clorados. Cada mezcla contiene desde el primero
hasta el último de los isómeros de esta clase de compuestos que se trabajan en
la columna DB-5 y SP 2331 (TABLA 17.7).
|
TCDD |
1,3,6,8 |
1,2,8,9 |
2,3,7,8 |
1,2,3,7 |
|
|
1,2,3,8 |
1,2,3,4 |
1,2,3,9 |
1,4,7,8 |
|
PeCDD |
1,2,4,6,8 |
1,2,3,8,9 |
|
|
|
HxCDD |
1,2,3,4,6,9 |
1,2,3,4,7,8 |
1,2,3,4,6,8 |
1,2,3,4,6,7 |
|
HpCDD |
1,2,3,4,6,7,8 |
1,2,3,4,6,7,9 |
|
|
|
OCDD |
1,2,3,4,6,7,8,9 |
|
|
|
|
|
|
|
|
|
|
TCDF |
1,3,6,8 |
1,2,8,9 |
|
|
|
PeCDF |
1,3,4,6,8 |
1,2,3,8,9 |
|
|
|
HxCDF |
1,2,3,4,6,8 |
1,2,3,4,8,9 |
|
|
|
HpCDF |
1,2,3,4,6,7,8 |
1,2,3,4,7,8,9 |
|
|
|
OCDF |
1,2,3,4,6,7,8,9 |
|
|
|
|
|
|
|
|
|
La
mezcla de evaluación de la columna se puede adquirir en Brem Laboratory, Wright
State University Dayton, Ohio, o equivalente.
8. Recolección, preservación y almacenamiento
de muestras
8.1 Cantidad de
muestra.
8.1.1 Tiempo de
muestreo-El tiempo de muestreo debe ser suficiente para ser representativo de
la fuente en cuestión. Por lo tanto no debe ser nunca menor a 3 h.
8.1.2 Volumen de
muestra-El volumen de muestra debe ser suficiente para reunir los analitos en
concentraciones mayores al límite de detección del método y las concentraciones
mínimas requeridas en la fuente en cuestión. Esto puede calcularse por la
siguiente fórmula:
Ecuación 3:
Volumen de Muestra (m3) = A x (100 / C) x (1 / D) x 3
donde:
A = Límite de detección del método analítico en ng/muestra (LDM)
obtenido en el laboratorio, ver Tabla 17.2.
C = % de Recuperación de la muestra (%R).
D = Límite Máximo Permitido (LMP) en la Fuente de Emisión (ng/m3).
Ejemplo:
LMP = 0.5 ng/m3, LDM = 1000 pg/muestra EQT (1.0 ng/muestra EQT), %R = 85%,
entonces:
V = 1.0 * (100/85) * (1/0.5) * 3 = 7.05 m3
Este
es el volumen mínimo que se debe tomar para asegurar que se tiene una detección
dentro del Límite Práctico de Cuantificación aun cuando la emisión está por
debajo de 3 veces el LMP.
8.2 El
único tratamiento especial en campo es el proceso de recuperación de la
muestra, el cual debe ser realizado bajo estrictas medidas de supervisión para
evitar la contaminación de los enjuagues y de las muestras de campo.
8.3 Las
muestras y enjuagues deben refrigerarse a 4°C desde el momento de su colecta
hasta el momento de la extracción.
8.4
Todas las muestras y enjuagues deben extraerse dentro de los 14 días
posteriores a su colecta y ser completamente analizadas dentro de los 45 días
después de la extracción.
9.
Control de Calidad
9.1
Muestreo
La identificación positiva y la cuantificación
de las PCDDs y PCDFs son altamente dependientes de la integridad de las
muestras recibidas y de la precisión y exactitud de todos los procedimientos
analíticos empleados. Los procedimientos de CC descritos en esta sección son
utilizados para evaluar el desempeño del muestreo, concentración y purificación
de las muestras y análisis cuali y cuantitativo.
9.1.1
Blanco del tren-Al menos 10% de los trenes en cada serie de pruebas corridas y
un mínimo de uno de estos trenes debe ser un blanco de tren. Prepare y coloque
el blanco de tren en una forma idéntica a aquella descrita en la sección 11. El blanco del tren debe llevarse a
través de todos los pasos desde la preparación hasta la verificación de fugas.
Recupere el blanco del tren como se describe en la sección 11.2. La resina XAD-2 utilizada en este
blanco, debe ser adicionada con los estándares surrogados del método al igual
que la utilizada en el tren de muestreo. Una recuperación aceptable en el campo
debe estar en el intervalo de 60 y 140 por ciento.
Los límites aceptables para la recuperación de
los estándares surrogados será de 100 ± 40% del valor conocido. Las muestras
que presentan resultados de estándares surrogados fuera de estos límites deben
tener una relación de señal a ruido mayor que o igual a 10, de lo contrario el
procedimiento analítico debe repetirse en la porción guardada del extracto.
9.2
Análisis
Cada laboratorio que use este método requiere
operar un programa de control de calidad formal. Los requerimientos de control
de calidad mínimos de este programa consisten en una demostración inicial de la
capacidad del laboratorio (de acuerdo a la sección 10.2 y análisis continuos de las muestras adicionadas para evaluar y
documentar la calidad de los datos. El laboratorio debe mantener los registros
del desempeño para documentar la calidad de los datos que se generan. Los datos
de calidad continua, verificados, son comparados con los criterios de desempeño
establecidos para determinar si los resultados de los análisis cumplen los
requerimientos del método.
9.2.1
Verificación de la contaminación-Antes de procesar cualquier muestra, el
analista debe demostrar a través del análisis del blanco de reactivos que todo
el material de vidrio y los reactivos están libres de interferencias al límite
de detección del método de la matriz de interés. Cada vez que un grupo de
muestras es extraído o se cambian reactivos, un blanco de reactivos debe
procesarse como una medida de seguridad en contra de la contaminación del
laboratorio. Un blanco de reactivos debe correrse a lo largo de cada lote de
muestras. La corrida de un blanco de reactivos debe realizarse exactamente
igual que la de las muestras, adicionándose todos los estándares internos y de
recuperación y realizándose todos los pasos de limpieza que a las muestras.
9.2.2
Blanco de matriz-Porciones de la matriz de la muestra (resina y filtro) deben
sujetarse a la extracción y limpieza seguido por el análisis HRGC/LRMS. Debe
haber al menos un blanco de matriz por cada lote de resina y filtros
preparados.
9.2.3
Muestras adicionadas de laboratorio-El laboratorio debe adicionar un blanco de
reactivos con una mezcla de dioxinas y furanos nativos para obtener la
exactitud del procedimiento analítico completo. La señal de la adición del
laboratorio debe ser de al menos 5 veces mayor que la de fondo. Debe existir
una muestra adicionada por cada lote de extracción.
9.2.4
Estándares internos-Cada muestra es adicionada con una cantidad conocida de un
estándar interno marcado isotópicamente estable y estándares surrogados
(sección 7.3.21) antes de la
extracción y el análisis. Las recuperaciones obtenidas de cada uno de estos
estándares debe ser mayor a 60% y menor que 120% del valor esperado.
Si la recuperación del estándar interno está
fuera de estos límites de aceptación, se debe señalar en el reporte de
resultados.
NOTA: Debido a que este
método es por dilución isotópica, cuando se aplica apropiadamente, es
independiente de la recuperación de los estándares internos. Recuperaciones
menores de los Estándares Internos no invalidan los resultados analíticos para
los PCDDs/PCDFs nativos detectados, pero puede resultar en límites de detección
más altos que los esperados.
Los
límites de acción para los resultados de los estándares internos serán de 60 a
120% del valor conocido. Las muestras que muestran resultados de estándares
internos fuera de estos límites deben tener una relación de señal a ruido mayor
que o igual a 10, de lo contrario el procedimiento analítico debe repetirse en
la porción guardada del extracto.
Cuando
estos procedimientos no producen resultados concluyentes, se sugiere la
utilización de Espectrometría de Masas de Alta Resolución o Espectrometría de
Masas.
9.2.5 Desempeño de las
Columnas DB-5 y SP-2331-El desempeño de las columnas cromatográficas debe
demostrarse inicialmente y verificarse cada 12 horas (sección 10.2). El desempeño de la columna debe
evaluarse con la solución de verificación de las columnas (7.3.23) bajo las mismas condiciones cromatográficas y de
espectrometría de masas utilizado en las muestras y los estándares. La
verificación consiste de la inyección de una mezcla conteniendo los isómeros
TCDD que eluyen cerca al 2,3,7,8-TCDD. Esta mezcla contiene 7 isómeros de TCDD
(2378, 1478, 1234, 1237, 1238, 1278 y 1267) incluyendo aquellos isómeros los
cuales se sabe son los más difíciles de separar en la columna SP 2331 y DB-5.
El
isómero 2,3,7,8-TCDD debe separarse del isómero interferente más cercano con
una resolución mayor a 25% en las dos columnas.
Los
siguientes isómeros deben resolverse en una columna de 60 m DB-5 con un valle
del 60% mínimo.
1,2,3,4,7,8-HxCDD
y 1,2,3,4,6,8-HxCDD.
9.2.6 Criterios de
identificación de PCDDs y PCDFs.
9.2.6.1 Todos los iones
característicos, tanto de cuantificación como de confirmación listados en la
Tabla 17.8 deben presentarse en un
cromatograma de iones reconstruido. Si se usa LRMS, el ión M-COCl debe
monitorearse también.
9.2.6.2 La intensidad
máxima de cada uno de los iones característicos especificados deben coincidir
dentro de 2 scans o 2 segundos.
9.2.6.3 El monitoreo de
la relación de masas debe estar dentro de ± 15% de la relación de masa estándar
especificado en la Tabla 17.8.
9.2.6.4 La señal para
medir la relación de ruido debe estar 2.5:1
o mayor para la cuantificación y confirmación de iones.
9.2.6.5 El tiempo de
retención de los congéneres nativos debe quedar dentro de ± 0.006 unidades del tiempo de retención
relativo de los estándares. Esta relación de los PCDDs/PCDFs nativos y de sus
estándares internos isotópicamente marcados debe mantenerse.
En
esta determinación, se sospechará la coelución de impurezas si todos los
criterios, excepto el criterio de relación isotópica, se cumple. Si se detectan
interferencias de fondo con picos anchos los cuales restringen la sensibilidad
del análisis de HRGC/LRMS, el analista debe emplear procedimientos adicionales
de limpieza y reanalizar el extracto purificado.
Cuando
estos procedimientos no arrojen resultados concluyentes, se sugiere el uso de
espectrometría de masas de alta resolución o espectrometría de masas-masas.
9.2.6.6 Para detecciones
confiables y cuantificación de PCDFs, se requiere que el analista monitoree las
señales que surjan de los policlorodifenil éteres, los cuales, si se presentan,
pueden dar iones de fragmentación con masas idénticas a aquellos monitoreados
como indicadores de PCDFs.
La
masa M+70 debe monitorearse para detectar a los policlorodifenil éteres
simultáneamente con la masa del ión PCDF (M). Sólo cuando la respuesta de la
masa del ión policlorodifenil éter no se detecta al mismo tiempo que la masa
del ión PCDF, puede considerarse la señal obtenida por el aparente PCDF como
única. Las interferencias positivas por los policlorodifenil éteres deben ser
reportadas o eliminadas.
9.2.6.7 Cuando se detecte
una posible PCDDs/PCDFs que cumple con todas las características anteriores,
como una confirmación adicional se debe reanalizar el extracto por medio de la
otra columna recomendada (si se utiliza DB-5 en el análisis se debe confirmar
con SP23331 o viceversa).
9.2.7 Prácticas de CC
adicionales-Se recomienda que el laboratorio adopte prácticas adicionales de CC
para usarse con este método. Las prácticas específicas que son más productivas
dependerán en algún grado en la naturaleza de las muestras.
9.2.7.1 Los
duplicados de campo deben analizarse para monitorear la precisión de las
técnicas de muestreo.
9.2.7.2
Cuando hay dudas acerca de la identificación de un pico en el cromatograma, deben
usarse técnicas confirmativas tales como la dilución de la muestra y la
fortificación de la misma.
9.2.7.3
Siempre que sea posible, el laboratorio debe analizar muestras de verificación
de control de calidad y participar en estudios de evaluación del desempeño con
muestras externas.
En vista a los rápidos avances que ocurren en
la cromatografía, se le permite al analista ciertas opciones para improvisar
separaciones o mediciones menos costosas. Cada vez que tales modificaciones son
hechas al método, el analista debe repetir los procedimientos descritos en las
secciones 9.2 y 10.2 y demostrar la habilidad para
generar datos con una precisión y exactitud aceptables.
9.2.8 Los
resultados y el procedimiento utilizado deberán quedar asentados en la bitácora
del analista y en el Expediente de la Demostración Inicial de Desempeño del
Método (EDIDM).
9.3
Límites de detección
9.3.1 En
el caso de que sean detectados PCDDs/PCDFs nativos, el límite de detección
actual debe estimarse y reportarse basado en una relación señal-ruido de 2.5:1 a un intervalo de masa apropiado.
Mida la media del ruido para el intervalo del tiempo de retención de cada
congénere del cromatograma de masas. Multiplique el ruido por 2.5 y calcule el límite de detección de
acuerdo a la ecuación de la sección 12.
9.3.2 Si
se presenta una señal interferente en el intervalo de masas, seleccione el ión
que no interfiera con el límite de detección calculado usando la ecuación de la
sección 12.0. Si ambos iones tienen
interferencias las cuales son mayores a 2.5
veces el ruido, calcule el límite de detección usando la masa que dará el
resultado más conservador.
9.4
Validación de modificaciones del método o de métodos alternos-Para validar las
modificaciones que se efectúen a este método o para la utilización de métodos
alternos deberá seguirse el siguiente procedimiento:
9.4.1 Si
se realizan modificaciones al presente método, deberán validarse de acuerdo a
lo que se presenta en la Sección 9.2.
9.4.2 Si
se utiliza un método alterno cuya fuente sea un método estandarizado por alguna
Institución de carácter internacional o reconocida internacionalmente (e. g.
ASTM, USEPA, AOAC, Standard Methods, DIN, OMS Environment Canada, etc.) siga el
mismo procedimiento que se presenta en la Sección 9.2.
9.4.3 Si
se utiliza algún método no estandarizado, deberá evidenciarse, además de los
parámetros mencionados en la Sección 9.2,
los parámetros de Robustez, Reproducibilidad y Especificidad, los cuales sólo
pueden evaluarse mediante estudios interlaboratorio.
10.
Calibración
10.1
Muestreo.
10.1.1
Boquilla de la Sonda-La boquilla de la sonda debe calibrarse de acuerdo al
procedimiento que se describe en la sección 5.1 de la CARB Método No. 5.
10.1.2 Tubo
Pitot-El procedimiento para calibrar el tubo Pitot tipo “S” es obtenido en la
sección 4 de la CARB Método No. 2.
10.1.3
Sistema de Medición-La calibración del sistema de medición debe ser optimizado
y realizado conforme a los requerimientos de la sección 5.3 de la CARB Método No. 5.
10.1.4
Termómetros (Indicadores de Temperatura)-Termómetros digitales, tales como los
usados en el equipo de muestreo isocinético y a la salida del condensador,
deben ser calibrados y comparados con termómetros de mercurio calibrados.
10.1.5
Verificación de las fugas del sistema de medición-La comprobación debe ser
realizada según lo establecido en la sección 5.6 de la CARB Método No. 5.
10.1.6
Barómetro-Debe calibrarse contra un barómetro de mercurio.
10.2
Calibración del Método Analítico.
Se debe analizar una curva de calibración por
la técnica de Estándar Interno.
10.2.1 Se
requieren dos procedimientos de calibración: La calibración inicial, la cual se
requiere antes de analizar alguna muestra y la calibración continua, la cual se
realiza antes y durante la corrida analítica, y la cual indica cuando se debe
recalibrar por el primer método el equipo, si no cumple los requisitos
especificados.
10.2.1.1
Calibración Inicial-Usando una solución Stock de estándares, prepare una curva
multinivel con las concentraciones que se presentan en la Tabla 17.3.
Se pueden inyectar 2.0 µL de los estándares de calibración por la técnica de inyección
splitless, asegúrese que se inyecta la misma cantidad de los estándares,
extractos de muestra y blancos.
Al establecer los parámetros de operación para
el sistema HRGC/LRMS, el instrumento debe sintonizarse para que cumpla los
criterios de la relación isotópica listados en la tabla 17.8 para las PCDDs y PCDFs. Una vez sintonizadas las masas y el
procedimiento de calibración se ha completado, una mezcla de chequeo debe
inyectarse al sistema HRGC/LRMS.
10.2.2
Calibración de la columna del Cromatógrafo de Gases:
Se deberá utilizar la mezcla de la
verificación de la columna (7.3.23),
el uso de esta mezcla de chequeo debe cubrir los siguientes parámetros:
10.2.2.1
Tiempo de retención para cada uno de los homólogos.
10.2.2.2 La
resolución entre la 2,3,7,8-TCDD y 1,2,3,4-TCDD.
10.2.2.3 Los
criterios de las abundancias relativas de los iones de las PCDDs y PCDFs en la
Tabla 17.9.
Debe haber al menos un valle de 25% entre las
masas cromatográficas de los picos observados para la 2,3,7,8-TCDD y los picos
adyacentes a otros isómeros de TCDD.
Dibuje la línea base para los isómeros
representativos 1,4,7,8-; 2,3,7,8-; 1,2,3,8-; 1,2,3,7- y 1,2,3,4-TCDD. Mida la
distancia x para la línea base al valle siguiente del pico de la 2,3,7,8-TCDD y
el pico alto de la 2,3,7,8-TCDD.
Ecuación 4: % Valle = (x / y) X 100
Es responsabilidad del analista verificar las
condiciones para alcanzar la máxima resoluciónde la 2,3,7,8-TCDD de los otros
isómeros de TCDD. El pico de la 2,3,7,8-TCDD debe etiquetarse como tal en todos
los cromatogramas.
Los siguientes compuestos pueden resolverse en
una columna de DB-5 de 60 m con un valle de un 60%: 1,2,3,4,7,8-HxCDD
y 1,2,3,4,6,8-HxCDD.
10.2.3
Sensibilidad del LRMS:
La sensibilidad del LRMS debe verificarse
durante la calibración inicial tomando como referencia una relación señal/ruido
de 5:1 para los iones de cuantificación obtenidos de la inyección del estándar
más bajo de la curva de calibración.
10.2.4
Factores de Respuesta Relativos:
De la inyección de los estándares de la curva
de calibración calcule los RRFs de las PCDDs/PCDFs nativos versus el apropiado
estándar interno.
Las dioxinas y furanos nativos y los
correspondientes estándares internos y los estándares utilizados para la
calibración se listan en la Tabla 17.9.
NOTA: Los
factores de respuesta relativos para congéneres de una serie homóloga deben ser
iguales. Por lo tanto, se asume que los factores de respuesta son los mismos.
Para minimizar los efectos de este concepto, un isómero sustituido disponible
comercialmente puede seleccionarse para cada serie 2,3,7,8 sustituido de las
PCDDs. Todos los RRF calculados para una serie homóloga se basan sobre este
compuesto en el caso de los penta a octa PCDDs, se asume que las respuestas
para los PCDFs son equivalentes a las de las PCDDs.
Para cada analito debe calcularse el Factor de
Respuesta Relativo contra su estándar interno respectivo (ver Tabla 17.6).
El Laboratorio debe demostrar que los RRF para
los Furanos y Dioxinas nativos son constantes.El promedio de los RRF debe
usarse para los cálculos. El % RSD de los RRF no debe exceder el 15% en el
rango de trabajo de la curva de calibración.
Los RRF deben verificarse para cada trabajo o
al menos cada 12 horas, por la medición de uno o más estándares de calibración
(si sólo se calcula uno debe ser en el rango medio de la curva de calibración).
Si las diferencias de respuestas calculadas exceden el 30%, debe prepararse una
nueva calibración.
Los RRF de los estándares surrogados deben
determinarse del mismo paquete de estándares los cuales contienen una cantidad
constante de estándares surrogados. Los RRF deben también verificarse cada 12
horas de trabajo. Si la respuesta varía más de 30% del factor de respuesta
establecido previamente, la prueba debe repetirse.
10.2.5
Calibración Rutinaria.
10.2.5.1
Evaluación rutinaria de la columna: Inyecte una alícuota de 2 µL de la mezcla
de evaluación de la columna (7.3.23).
Obtenga al menos 5 espectros para cada pico cromatográfico.
NOTA: Los
mismos parámetros de adquisición de datos previamente usados para analizar la
concentración de las soluciones de calibración deben usarse para la solución de
evaluación del sistema. La solución de evaluación del sistema debe correrse al
inicio y al final de cada periodo de 12 horas. Si el laboratorio opera durante
12 horas consecutivas, es suficiente el análisis de la mezcla de evaluación del
sistema al principio y al final.
Documente la adecuada operación de la columna.
10.2.5.2
Evaluación de la calibración inicial: Inyecte una alícuota de 2 µL del punto de
200 pg/µL de la mezcla de calibración y determine que la variación de la
respuesta sea menor de 30% respecto a la calibración original, en caso de que
no sea así, repita la calibración inicial.
11.
Procedimiento
11.1
Muestreo-Debido a la complejidad del método, se requiere que los muestreadores
estén entrenados y tengan amplia experiencia para asegurar la veracidad y
reproducibilidad de los resultados.
11.1.1
Pruebas preliminares obligatorias-Todos los componentes deben calibrarse de
acuerdo a los procedimientos APTD-0576 o una calibración similar.
Pese varias porciones de 200 a 300 g de sílica
gel en contenedores herméticos con una precisión de 0.5 g o como una alternativa pese directamente en los impactores
que serán utilizados en el tren de muestreo un poco antes de utilizarse.
Revise los filtros visualmente a contra luz
para detectar fallas en el filtro y prevenir fugas. Etiquete los frascos que
contendrán los filtros y guarde en ellos los filtros pesados.
Estabilice los filtros a 20 ± 5.6°C a presión atmosférica durante 24
horas pesando los filtros cada 6 horas para asegurarse que se ponen a peso
constante o sea que no deberán variar más de 0.1 mg de la última pesada. Durante las pesadas nunca deberán
exponerse los filtros por más de 2 minutos a una humedad relativa mayor a 50%.
Alternativamente los filtros pueden secarse en un horno a 105°C por 3 horas y
guardarse en desecador.
11.1.2 Determinaciones
preliminares-Seleccione el sitio de muestreo y el número de puntos de muestreo
de acuerdo al procedimiento descrito en el Método CARB No. 1.
Determine la presión de la chimenea,
temperatura y velocidad de los gases, de acuerdo al procedimiento descrito en
el Método CARB No. 2, verifique las fugas en las líneas del tubo Pitot.
Determine el contenido de humedad utilizando
el procedimiento descrito en el Método CARB No. 4.
Determine el peso molecular del gas de la
chimenea en base seca de acuerdo al procedimiento descrito en el Método CARB
No. 2.
Seleccione el tamaño de la boquilla según la
velocidad de los gases, de tal manera que no sea necesario algún cambio de
boquilla para poder mantener el muestreo isocinético. Durante la corrida no
cambie el tamaño de la boquilla. Asegúrese que la presión diferencial sea
adecuada y seleccione el rango de velocidades que se encontrarán en la chimenea
durante la corrida (ver Método CARB No. 2).
Seleccione la longitud adecuada de la sonda de
muestreo, de manera que todos los puntos transversales puedan muestrearse. Para
muestrear chimeneas grandes considere el muestreo por los lados opuestos para
poder determinar el número suficiente de puntos transversales.
El tiempo total de muestreo debe ser mayor o
igual al tiempo mínimo para muestrear el volumen requerido en la sección 8.1.
Se recomienda que el tiempo en cada punto
transversal muestreado sea al menos de 2.0
minutos.
11.1.2.1
Limpieza del Material-Todas las partes de vidrio del tren de muestreo, incluido
el módulo de la resina y los impactores deben lavarse como se especifica en la
Sección 3 A del Manual de Métodos Analíticos para Análisis de Residuos de
Plaguicidas en Muestras Ambientales y Humanas de la USEPA. Debe tenerse
especial cuidado en la limpieza de las partes que tuvieron grasa de silicona en
las conexiones de vidrio. Los residuos de grasa deben removerse completamente
por el remojo durante varias horas con mezcla crómica antes de la rutina de
limpieza mencionada.
Todo el material de vidrio debe enjuagarse con
cloruro de metileno antes de usarse en el tren de muestreo de PCDDs/PCDFs.
11.1.2.2
Módulo de la Resina XAD-2-Use una cantidad suficiente (al menos 30 g o 5
gramos/m3 del
gas a muestrearse) de resina XAD-2 limpia para llenar completamente el módulo
contenedor el cual ha sido previamente limpiado y enjuagado con hexano. La
trampa y la lana de vidrio deben enjuagarse en repetidas ocasiones con los
disolventes que se utilizan para lavar la resina. El contenedor de la resina no
debe destaparse hasta que se va a montar el tren de muestreo.
La adición de los estándares surrogados debe
ser realizada en el laboratorio de 6 a 12 horas antes de iniciar el muestreo.
11.1.3 Si
se usan impactores para condensar la humedad de los gases de la chimenea
prepárelos de la siguiente manera: Ponga 100 mL de agua en el primer impactor y
100 mL de etilén glicol o agua en el segundo, en el tercer impactor ponga 250 a
300 gramos de sílica gel previamente pesada, anote en la bitácora el peso, ya
que se utilizará para el cálculo de la humedad por gravimetría.
NOTA: No
utilice grasas de silicón o de otra clase para sellar fugas.
Ponga el contenedor en un lugar limpio para
más tarde utilizarlo en la recuperación de la muestra.
Utilizando pinzas o guantes coloque el filtro
en el portafiltros asegurándose que quede centrado de manera que el empaque
quede sellado, revise el portafiltros y el filtro para detectar rasgaduras.
Ponga cinta resistente al calor alrededor de
la sonda para marcar los puntos transversales a muestrear.
Ensamble el tren de muestreo y coloque trozos
de hielo alrededor de los impactores.
11.1.4
Procedimiento de revisión de fugas.
11.1.4.1
Prueba previa de revisión de fugas.
Se requiere una verificación previa de fugas
según el siguiente procedimiento:
Después de que el tren de muestreo ha sido
ensamblado prenda el sistema de calentamiento de la sonda y espere a que esté a
las condiciones requeridas de operación, permita que la temperatura se
estabilice. Revise fugas en todas las uniones y conexiones del tren de muestreo
y conexiones de la boquilla por medio de la obturación de la boquilla con un
tapón de teflón limpio y aplique un vacío de 380 mm de Hg. (15”).
NOTA:
Puede usarse un vacío más bajo a condición de que en ningún momento de la prueba
se rebase tal vacío.
Una fuga en exceso de 4% del promedio del
rango de muestreo o 0.00057 metros
cúbicos por minuto es inaceptable.
Para verificar fugas en el tren de muestreo,
utilice el siguiente procedimiento: Inicie bombeandocon la válvula de “by-pass”
totalmente abierta y la válvula del ajuste grueso totalmente cerrada.
Parcialmente abra la válvula del ajuste grueso y lentamente cierre la de
“by-pass” hasta obtener el vacío adecuado. No regrese la válvula del “by-pass”
ya que podría regresar agua al portafiltro, si el vacío deseado se excede,
verifique las fugas a ese valor de vacío y termine de revisar las fugas como se
indica abajo y continúe.
Cuando se terminan de revisar las fugas
primero remueva lentamente el tapón que se colocó en la boquilla,
inmediatamente apague la bomba de vacío. Debe cuidarse que la sílica gel no se
humedezca.
11.1.4.2
Verificación de fugas durante el muestreo:
Si durante el muestreo un componente (ejemplo
el filtro, un impactor u otro ensamble) se cambia, deben verificarse las fugas
inmediatamente después de haber hecho el cambio. La verificación de las fugas
debe realizarse según la sección 11.1.4.1
excepto que deberá hacerse a un vacío igual o mayor al valor máximo registrado
en la prueba. Si la fuga detectada presenta un flujo menor a 0.00057 m3/min o 4% del
flujo promedio muestreado el resultado es aceptable sin necesidad de
corrección. Si la fuga es mayor a lo estipulado corrija el volumen total según
la sección 11.3 de este método o el
muestreo se debe anular.
11.1.4.3
Verificación de fugas posterior.
Es obligatoria una verificación de fugas al
concluir cada uno de los muestreos. La verificación de fugas deberá realizarse
según el procedimiento de la sección 11.1.4.1
excepto que debe conducirse a un vacío igual o mayor al máximo registrado
durante la prueba. Si el flujo de la fuga no es mayor a 0.00057 m3/min
o 4% del promedio del flujo muestreado el resultado es aceptable y no requiere
que se aplique la corrección al total del flujo muestreado. Si la fuga es mayor
corrija el volumen final según la sección 11.3
de este método o el muestreo se debe anular.
11.1.4.4
Corrección de una fuga excesiva.
El volumen muestreado obtenido mediante la
ecuación que se da en la sección 12.3
de este método, se corrige por medio de la ecuación 5 si excede (La) el máximo
aceptable de fugas, reemplace (Vm) en la ecuación 12.3 por:
Ecuación 5: Vm = (Li-La)Oi-(Lp-La)Op
donde:
Vm = Volumen de gas muestreado y
medido
La = Máximo aceptable de fugas 0.00057 metros cúbicos por minuto (0.02 cfm) o 4% del flujo promedio
muestreado.
Lp = Volumen de fuga observada
durante la verificación de fugas al final de la prueba.
Li = Volumen de fuga
observado durante la verificación realizada en los diferentes muestreos
(i=1,2,3,...n), metros cúbicos por minuto.
Oi = Tiempo de muestreo entre
dos sucesivas verificaciones de fugas, iniciando con el intervalo de primero y
el segundo.
Op = Tiempo de muestreo entre
la última verificación de fugas y la prueba final.
Substituya solamente los siguientes rangos (Li
o Lp) los cuales excedan (La).
11.1.5
Operación del Tren.
11.1.5.1
Durante el muestreo, mantenga el flujo dentro de un ± 10% del valor isocinético
real; para cada corrida registre la información requerida en la hoja de campo.
Asegúrese de registrar la lectura inicial del equipo. Registre las lecturas del
equipo al principio y al final de cada incremento de tiempo en el muestreo,
cuando se realice un cambio en el flujo, antes y después de cada verificación
de flujo y cuando se suspenda el muestreo.
11.1.5.2
Registre otras lecturas requeridas en la hoja de campo y llene una por cada
punto de muestreo transversal, por cada incremento significativo que tenga un
20% de variación en la velocidad, se requieren ajustes adicionales en los
flujos.
11.1.5.3
Nivele y ponga a cero el manómetro inclinado-El nivel y el cero del manómetro
pueden tener variaciones debido a vibraciones y variaciones de temperatura,
realice verificaciones periódicas.
11.1.5.4
Limpie los puertos de muestreo antes de la corrida de prueba para minimizar el
muestreo de los depósitos de material. Para iniciar el muestreo remueva la
boquilla y verifique que el tubo Pitot y la extensión de la sonda sea la
adecuada y que se encuentren bien colocadas. Coloque la boquilla en el primer punto
transversal con la punta directamente hacia el flujo de gas.
11.1.5.5
Inmediatamente empiece a bombear y ajuste el flujo a la condición isocinética.
Si se disponen de nomogramas, éstos sirven de ayuda para ajustar rápidamente el
muestreo isocinético.
11.1.5.6 Estos nomogramas
están diseñados para cuando se usan tubos Pitot tipo “S” con un coeficiente
(Cp) de 0.85 ± 0.02 y la densidad del gas equivalente de la chimenea (peso
molecular promedio en base seca) (Md) sea igual a 29 ± 4. Si Cp y Md están afuera de los rangos de inicio, no use el
nomograma sin que se tomen los pasos apropiados para compensar las
desviaciones.
11.1.5.7 Cuando las
chimeneas tienen una presión negativa significativa, ponga atención al cerrar
la válvula del ajuste grueso antes de insertar la sonda dentro de la chimenea
para prevenir que el agua se regrese. Si es necesario, la bomba puede prenderse
con la válvula del ajuste grueso cerrada, cuando la sonda esté en posición,
bloquee o tape alrededor de la sonda en el puerto de muestreo para que no haya
dilución de los flujos de gases.
11.1.5.8 Cuando se esté
muestreando la sección transversal de la chimenea debe tenerse cuidado de no
bombear cuando se está metiendo o ensamblando la boquilla en la sonda para no
remover material sólido de las paredes.
11.1.5.9 Durante la
corrida de prueba tome las precauciones necesarias para mantener la temperatura
del condensador por debajo de 20°C (adicione trozos de hielo al baño de hielo
del impactor). Esto ayuda a evitar la pérdida de humedad. También cheque
periódicamente el nivel y el cero del manómetro.
11.1.5.10 Si la caída de
presión aumenta en el filtro y esto impide la medición isocinética, reemplace
el filtro durante la corrida. Es recomendable se tenga preparado todo un nuevo
ensamble o portafiltro para usarse. Antes de operar el nuevo ensamble realice
una verificación de fugas. El total de las partículas será el reunido en los
dos filtros.
11.1.5.11 Debe utilizarse
un solo tren para obtener la muestra, excepto cuando se requieren muestreos
simultáneos de dos o más ductos separados o dos o más lugares diferentes del
mismo ducto.
11.1.5.12 Al final de la
corrida apague la bomba, remueva la sonda y registre la lectura final del
equipo. Realice una verificación de las fugas. Las líneas deberán pasar la
presente verificación de fugas para dar validez al trabajo.
11.1.6 Método de cálculo
del porcentaje de isocinetismo-Calcule el % de isocinetismo para determinar si
la prueba fue válida, en caso contrario se debe llevar a cabo una segunda prueba.
11.2 Recuperación de
la Muestra.
Al
final del muestreo es necesario limpiar la sonda tan rápido como sea posible
para remover la contaminación captada en la chimenea.
Cuando
la temperatura permita manejar la sonda con seguridad, limpie todo el exterior,
remueva la sonda del tren de muestreo y tápela con papel aluminio previamente
lavado con cloruro de metileno. Selle la entrada del tren con su tapón y papel
aluminio enjuagado con hexano.
Transfiera
la sonda y los impactores al área de recuperación previamente seleccionada,
tápese para asegurar que no se contamine ni se pierda la muestra. En el área de
recuperación de la muestra no se debe fumar.
Inspeccione
el tren de muestreo e investigue si hay alguna anormalidad, por ejemplo un
filtro roto, fuga de algún líquido, cambio de color, etc.
11.2.1
Contenedor No. 1-Cuidadosamente remueva el filtro del portafiltros y colóquelo
en el contenedor identificado como “Contenedor No. 1”. Utilice unas pinzas
previamente lavadas para manejar el filtro. Si es necesario doble el filtro,
asegurándose que las partículas queden dentro del mismo. Cuidadosamente
transfiera al contenedor las partículas y las fibras del filtro que se hayan
quedado en el contenedor del filtro. Para limpiar el contenedor del filtro
utilice un cepillo seco e inerte.
11.2.2
Módulo de la Resina-Remueva el módulo de la Resina XAD-2 del tren de muestreo y
tápelo con papel aluminio lavado con hexano.
11.2.3
Contenedor de muestra No. 2-Recupere cuantitativamente el material depositado
en la boquilla, en la sonda, en la línea de transferencia, en la parte frontal
del portafiltro y en el ciclón, si fue usado. Primero cepille y luego enjuague
secuencialmente tres veces con Metanol, Benceno y Cloruro de Metileno. Ponga
todos los enjuagues en el contenedor No. 2.
11.2.4
Contenedor de muestra No. 3-Enjuague la parte trasera del portafiltro, la
conexión entre la línea y el refrigerante (si se usó el condensador separado
del contenedor de la resina) tres veces secuencialmente con Metanol, Benceno y
Cloruro de Metileno y coléctelos en el contenedor No. 3. Si se usó una trampa/condensador, el enjuague de la trampa debe
realizarse en el laboratorio después de remover la porción de resina XAD-2. Si se usó una trampa expulsora de
agua, el contenido y los enjuagues deben ponerse en el contenedor No. 3. Enjuague 3 veces con Metanol,
Benceno y Cloruro de Metileno.
11.2.5
Contenedor de muestra No. 4-Remueva el primer impactor, seque y limpie la parte
exterior del impactor. El contenido y los enjuagues póngalos en el contenedor
No. 4. Enjuague el impactor
secuencialmente tres veces con Metanol, Benceno y Cloruro de Metileno.
11.2.6
Contenedor de muestra No. 5-Remueva el segundo y tercer impactor, seque la
parte exterior y limpie. Vacíe el contenido y los enjuagues dentro del
contenedor No. 5. Enjuague cada uno
con agua tres veces.
11.3
Análisis.
En los muestreos de chimeneas van a resultar
muestras líquidas y sólidas para su análisis. Las muestras deben combinarse
como sigue:
1) El
filtro y las partículas colectadas en el filtro (Contenedores No. 1 y No. 2).
2) El
contenedor de la muestra No. 3 y la resina y los enjuagues del cartucho de
resina.
3)
Contenedores de la muestra No. 4 y No. 5.
Es preferible que las muestras no se dividan
para su análisis, ya que es muy difícil obtenerlas homogéneas como generalmente
ocurre con las muestras líquidas. Las muestras sólidas tales como la resina no
es homogénea y el filtro es tan pequeño que el límite de detección
probablemente no se alcance, si se divide la muestra.
PRECAUCION: Cuando se utiliza este método
todas las operaciones deben realizarse en áreas restringidas a personas que
estén perfectamente entrenadas en las medidas de seguridad y de protección para
evitar exposiciones a la piel.
11.3.1
Extracción de Muestras Líquidas.
11.3.1.1
Contenedor de muestra No. 2.
Concentre los enjuagues del contenedor de
muestra No. 2 (sección 11.2.4) a un
volumen de 1 a 5 mL usando un Turbo-Vap a una temperatura de 50°C. El residuo
contendrá partículas que fueron removidas del tren de muestreo. Combine el
residuo (enjuague 3 veces con el contenido del recipiente final de la muestra)
en el Soxhlet con el filtro y las partículas y proceda como se indica en la
sección 11.3.2.1.
11.3.1.2
Contenedor de muestra No. 3.
Concentre los enjuagues del contenedor de
muestra No. 3 (sección 11.5.5) a un
volumen de 1 a 5 mL usando un Turbo-Vap a 50°C. Concentre casi a sequedad.
Combine los residuos (con tres enjuagues del recipiente de muestra final) en el
soxhlet con la muestra de resina y proceda como se describe en la sección 11.3.2.1.
11.3.1.3
Contenedor No. 4 y No. 5.
Combine el contenido del frasco No. 4 y No. 5
(sección 11.2.5 y 11.2.6) en un embudo de separación.
Extraiga la muestra tres veces con tres porciones de Cloruro de Metileno. Combine
la fracción orgánica en un matraz que contenga Na2SO4 anhidro. Adicione
500 µL de tetradecano y concentre a 500 µL en un Kuderna-Danish o rotavapor y
transfiera el extracto a un tubo de prueba de 8 mL con hexano. Combine el
extracto en el Soxhlet con las muestras sólidas como se describe en la sección
11.3.2.1.
11.3.2
Extracción de Muestras Sólidas.
11.3.2.1
Filtro y Partículas Retenidas.
El Soxhlet debe limpiarse por 8 horas mínimo
con el disolvente de extracción y el disolvente debe descartarse. Adicione 20
gramos de Na2SO4 en el cartucho.
Corte el filtro en pequeños trocitos y
póngalos en el lugar de la muestra junto con los enjuagues (sección 11.3.1.1 a 11.3.1.3) sobre la parte alta del sulfato de sodio anhidro, agregue
la resina XAD-2, ponga un tapón de fibra de vidrio lavada y adicione 50 µL de
los estándares internos isotópicamente marcados.
Ponga el cartucho en el Soxhlet y adicione 250
mL de Tolueno al matraz. Ensamble el Soxhlet, prenda la parrilla de
calentamiento y abra la llave del agua del refrigerante y refluje durante 16 h.
Después de la extracción, permita que el Soxhlet se enfríe. Transfiera a un
matraz de 500 mL y adicione aproximadamente 500 µL de tetradecano. Adicione
aproximadamente 50 mL de Hexano y concentre a un volumen de 500 µL en un
Kuderna-Danish o Turbo-Vap. Transfiera el extracto a un tubo de prueba de 8 mL
con hexano y guarde el extracto para su limpieza en columna.
Limpieza Opcional Preliminar.
Ciertas muestras que se encuentran muy sucias
pueden requerir una limpieza preliminar antes de ser cromatografiadas. En tal
caso, se debe seguir el procedimiento siguiente: Lave el extracto orgánico con
25 mL de agua agitando 2 minutos, permita reposar para que se separen las
fases, descarte la fase acuosa y la fase orgánica póngala en un matraz
Erlenmeyer.
PRECAUCION: Adicione 50 mL de ácido sulfúrico
concentrado al extracto orgánico, agite por 10 minutos. Permita que la mezcla
se separe en un embudo de separación (aproximado 10 min). Descarte con mucho
cuidado la fase acuosa/ácida. Repita la operación hasta que el ácido casi no
presente coloración.
Ponga la muestra en un embudo de separación y
adicione 25 mL de agua, agite 2 minutos y permita separar las fases. Descarte
la capa acuosa y seque la capa orgánica con sulfato de sodio anhidro.
Transfiera el extracto orgánico a tubos de
Turbo-Vap y evapore a 55°C casi a sequedad.
Reconstituya en hexano antes de proceder con
la columna cromatográfica.
11.3.3
Columnas de Limpieza.
El extracto obtenido como se describe en la
sección anterior es concentrado a 1 mL utilizando el Turbo-Vap. Transfiera
cuantitativamente con tres enjuagues de 1 mL a la columna de sílica gel/alúmina
como se describe abajo.
11.3.3.1
Preparación de Columna.
A.
Columna combinada de Sílica gel/Alúmina-Se empaca por gravedad una columna de
200 mm por 15 mm de la siguiente manera:
Lana de vidrio Inerte (Silanizada) se coloca
en la punta de la columna y adicione, en secuencia, 1 g de sílica gel, 2 g de
sílica gel modificada básica, 1 g de sílica gel, 4 g de sílica gel modificada
ácida, 1 g de sílica gel y una capa de 1 cm de sulfato de sodio anhidro.
B.
Columna ácida de Alúmina-Empaque por gravedad una columna de vidrio de 11 mm de
diámetro, como sigue:
Coloque lana de vidrio Inerte (Silanizada) en
la punta de la columna. Adicione 6 g de alúmina ácida preparada como se
describe en la sección 7.3.14 golpee
suavemente la columna hasta que se asiente la alúmina y adicione 1 cm de
sulfato de sodio anhidro.
C.
Columna de Carbopak-Celite-Tome una pipeta serológica de 5 mL y corte 1 cm de
la punta, ponga lana de vidrio silanizada y lavada con cloruro de metileno.
Adicione suficiente Carbopak-Celite (0.3
g) a la columna hasta hacer 2 cm de longitud y coloque un tapón de lana de
vidrio en la parte superior.
11.3.3.2 Procedimiento
de Limpieza.
Eluya las columnas “A” y “B” con hexano y
descarte el eluato. Cheque que la columna no tenga canales, si tiene
descártela. No tape la columna húmeda.
Adicione el extracto de muestra con 5 mL de
hexano a la parte alta de la columna “A” seguido de dos porciones de 5 mL de
hexano para enjuagar. Eluya la columna “A” con 90 mL de hexano directamente
sobre la columna “B”. Eluya la columna “B” con 20 mL de hexano/cloruro de
metileno al 20% volumen. Concentre el extracto a 0.50 mL utilizando el Turbo-Vap.
NOTA: La
concentración óptima de cloruro de metileno va a variar con la actividad de la
alúmina. En cada lote de alúmina el analista debe determinar la concentración
óptima para eluir las bajas concentraciones de los estándares de calibración
sin eluir las interferencias de la columna.
Eluya la columna C con 5 mL de hexano en un
sentido y luego en el sentido inverso del flujo. Mientras está en el sentido
inverso, eluya con 2 mL de tolueno, 1 mL de cloruro de metileno/metanol/benceno
(75/20/5) v/v, 1 mL de cloruro de metileno/ciclohexano (50/50 v/v) y 2 mL de
hexano. Descarte los eluatos.
Mientras esté el flujo en la dirección
inversa, transfiera el concentrado de la muestra a la columna con hexano y
eluya la columna en secuencia con 1 mL de hexano, 1 mL de cloruro de
metileno/ciclohexano (50/50 v/v) y 1 mL de cloruro de metileno/metanol/benceno
(75/20/5 v/v). Descarte el eluato. Dé la vuelta a la columna y eluya con 4 mL
de tolueno. Guarde este eluato para el análisis de las PCDDs/PCDFs. Evapore la
fracción del tolueno a 1 mL aproximado en un Turbo-Vap a 50°C.
Transfiera
a un microvial usando enjuagues de tolueno y concentrando a 50 L usando un flujo
de nitrógeno extra seco. Almacene los extractos en un congelador tapándolos de
la luz hasta que se vayan a analizar por GC/MS.
NOTA: Los extractos de
las PCDDs/PCDFs son muy sensibles a la luz del sol, deben ser guardados en
viales ámbar y protegidos con papel aluminio de la luz en general, bajas
recuperaciones se obtendrán si no se protegen de la luz los extractos.
11.4 Análisis por
GC/MS.
11.4.1 En la Tabla 17.7 se encuentran las columnas
capilares cromatográficas típicas y las condiciones de operación. Las
condiciones del HRGC deben establecerse por cada analista y para cada
instrumento usado. Deben inyectar alícuotas de las mezclas de chequeo. El
analista podrá hacer ajustes ligeros de las condiciones de operación sobre del
análisis de las mezclas.
11.4.2 Aproximadamente
una hora antes del análisis por HRGC/LRMS, ajuste el volumen del extracto a
aproximadamente 40 µL. Se deben agregar en este momento 10 µL de la solución de
estándares de recuperación (sección 7.3.22).
11.4.3 Analice las
muestras y estándares utilizando el LRMS en la modalidad de SIM para obtener al
menos 5 puntos por pico. Use exactamente las masas de la Tabla 17.8 hasta un décimo para las tetras,
pentas, hexas, heptas y octa isómeros de las dioxinas y su apropiado estándar
interno.
11.4.4 El sistema debe
calibrarse diariamente como se describe en la sección 10.0. El volumen del estándar de calibración inyectado debe ser el
mismo que las muestras.
11.4.5 Inyecte 2 µL de
muestra en la columna DB-5.
11.4.6 La presencia de
congéneres de tetra a octa deben ser cualitativamente confirmados.
11.4.7 Para la
cuantificación, se mide la respuesta de la masa del congénere nativo y del
estándar interno (Tabla 17.8). Una
corrección debe hacerse por contribución a la masa m/e 328 por alguna TCDD
nativa si ésta estuviera presente. Para hacer esto reste 0.009 por la respuesta del ión 322 a la respuesta del ión 328.
Calcule
la concentración de los congéneres nativos usando el factor de respuesta
relativo (RRF) y la ecuación 6. Si
al calcular la concentración está por arriba del punto más alto del rango de
calibración, reporte con la nota apropiada, que los datos fueron obtenidos por
extrapolación de la curva de calibración. La muestra debe ser diluida y
reinyectada sólo en caso de que el detector se haya saturado. Si los congéneres
nativos no están presentes, calcule el límite de detección.
La
concentración de los isómeros 1,2,3,4,6,7,8-HpCDD, 1.2.3.4.6.7.8-HpCDF, 1,2,3,4,7,8,9-HpCDF y OCDF se calcula a partir
de los resultados obtenidos en la columna DB-5.
La
concentración de la 2,3,7,8-TCDD, 2,3,7,8 TCDF, 1,2,3,7,8-PeCDD y PeCDF, 2.3.4.7.8-PeCDF, 1,2,3,4,7,8-HxCDD y
HxCDF, 1,2,3,6,7,8-HxCDD y HxCDF, 1,2,3,7,8,9-HxCDD y HxCDF y 2,3,4,6,7,8-HxCDF
se calculan a partir de los resultados obtenidos en la Columna de SP2331.
11.5 Análisis
Cualitativo:
11.5.1 Ventana de
Tiempos de Retención: La ventana de tiempos de retención para una serie
homóloga se define como el periodo de elución de un grupo de congéneres,
empezando en el punto donde el primer congénere eluye y terminando donde eluye
el último congénere. Estas ventanas deben ser establecidas en el montaje del
método y no deben de traslaparse entre los diferentes tetra, penta, hexa, hepta
y octa cloro dioxinas o furanos.
Todos
los picos que aparezcan dentro de las ventanas definidas son sospechosos de ser
PCDDs/PCDFs.
11.5.2 Todos los iones
característicos (el de cuantificación y el de confirmación) deben estar
presentes en el cromatograma reconstruido, la relación entre estos iones debe
cumplir con un ± 15% de los valores de la Tabla 17.8.
11.5.3 La intensidad
máxima de los iones característicos y de confirmación deben coincidir dentro de
un rango de 2 scans o 2 segundos.
11.5.4 Tiempos de
retención relativos: Los TRR del congénere nativo con su correspondiente
isótopo marcado debe coincidir dentro de un rango de ± 0.006 TRR.
11.5.5 Relación señal a
ruido: La relación señal a ruido promedio debe ser al menos de 2.5.
12. Cálculos
Realice
todos los cálculos al menos con una cifra decimal más de los datos obtenidos.
Redondee los cálculos en el resultado final.
12.1
Nomenclatura
An = Area seccional
Cruzada de la boquilla (m2).
Bws = Vapor de agua en
el gas, proporción por volumen.
Cs = Concentración
de PCDDs/PCDFs en gases de chimenea, ng/dscm, corregida a condiciones estándar
de 20°C, 760 mm Hg. (68°F, 29.92 in
Hg.) en base seca.
Gs = Masa Total de
PCDDs/PCDFs en muestras de gas en chimenea, en ng.
I = % de
isocinetísmo en el muestreo.
La = Rango máximo
aceptable de fuga para cada uno de las pruebas previas de verificación de fugas
o para la verificación de fugas seguido del cambio de un componente;
equivalente a 0.00057 m3/min
o 4% del promedio del rango de muestreo, cualquiera que sea el menor.
Li = El rango de
fugas observado durante la verificación de fugas realizado antes del tercer
cambio de componente (I=1,2,3,....n) m3/min.
Lp = El rango de
fugas observado durante la verificación posterior a la toma de muestra.
M = Peso
molecular del agua, 18.0 g/g-mol.
Pbar = Presión
barométrica del sitio de muestreo, mm Hg.
Ps = Presión
absoluta de la chimenea, mm Hg.
Pstd = Presión estándar
absoluta, 760 mm Hg.
R = Constante
ideal del gas 0.06236 mm Hg-m3 /
°K -g-mole.
Tm = Promedio de la
temperatura absoluta del gas seco (°K).
NOTA: Tm va a depender del tipo de medidor usado en la configuración del
muestreo.
Ts = Promedio de la
temperatura absoluta del gas de la chimenea (°K).
T std = Temperatura
estándar absoluta (293°K).
Vaw = Volumen de
acetona utilizado en el lavado, mL.
V1c = Volumen total de líquido colectado en el
impactor y sílica gel, mL.
Vm = Volumen del gas
muestreado y medido con el equipo muestreador (m3).
Vm(std) = Volumen de gas
muestreado y medido por el equipo muestreador, corregido por las condiciones
estándar (dsm3).
Vs = Velocidad del
gas de la chimenea, calculada por el Método CARB No. 2, ecuación 2-9,
utilizando los datos obtenidos del Método CARB No. 5 (m/seg).
Y = Factor de
calibración del medidor de gas seco.
?H = Promedio de la
presión diferencial del orificio transversal (mm H2O).
O = Tiempo total
muestreado en minutos.
O1 = Intervalo
de tiempo muestreado, desde el inicio de la corrida hasta el primer cambio de
componentes, en minutos.
Ol = Intervalo
de tiempo entre el cambio del primer componente y el segundo, en minutos.
Op = Intervalo
de tiempo, del cambio del componente final (nth) hasta el final de la corrida,
en minutos.
13.6 = Gravedad
específica del mercurio.
60 = seg/min.
100 = Conversión
a por ciento.
12.2
Promedio de temperatura del medidor de gas seco y promedio de la caída de
presión del orificio, ver los datos de la hoja de campo.
12.3
Volumen del gas seco-Volumen de muestra corregido medido por el medidor de gas
seco a condiciones estándar (20°C, 760 mm Hg). Utilizando la ecuación 6.
Ecuación
6:
![]()
donde:
![]()
NOTA: La
ecuación 6 puede ser usada como está escrita, si las fugas observadas durante
alguna de las verificaciones obligatorias (ejemplo: la verificación de fugas
posterior a la prueba o realizados por concepto de cambio de componentes)
exceden La o si Lp o L1 excede la ecuación 6 debe ser modificada
como sigue:
(a) Caso
No. 1 Si no hay cambio de componentes durante la corrida de toma de muestra. En
este caso reemplace Vm en la ecuación 6 con la expresión:
Ecuación
8: Vm = ( Lp -La)
O
(b) Caso
2 Si se cambian uno o más componentes durante la corrida de toma de muestra. En
este caso, reemplace Vm en la ecuación 6 con la expresión:
![]()
y substituya solamente por las fugas (Li o Lp) que excedan a La.
12.4
Factores de conversión.
|
De |
a |
Multiplicar
por |
|
scf |
m3 |
0.02832 |
|
g/ft3 |
gr/ft3 |
15.43 |
|
g/ft3 |
lb/ft3 |
2.205 * 10-3 |
|
g/ft3 |
g/m3 |
35.31 |
12.5 Variación
Isocinética.
12.5.1
Cálculo a partir de los datos crudos:
donde:
K3 = 0.003454
mm Hg-m3/mL-°K.
12.5.2
Cálculo a partir de valores intermedios:
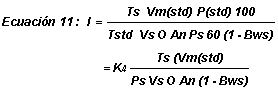
donde:
K4 = 4.320
12.6
Resultados aceptables: Si 90% < I <110 por ciento los resultados son
aceptables. Si se encuentra un sesgo o desviación en el resultado por ejemplo I
< 90%, quiere decir que el resultado es menor al valor determinado y puede o
no aceptarse. Si el resultado presenta un sesgo o desviación I > 110%,
quiere decir que el valor es mayor al valor determinado y puede o no aceptarse
el resultado.
12.7
Concentración de las PCDDs/PCDFs en los gases de la chimenea-Determine la
concentración de PCDDs/PCDFs en gases de chimenea de acuerdo con la siguiente
ecuación:
![]()
12.8
Concentración de isómeros individuales-La concentración de los isómeros
individuales de tetra, penta, hexa, hepta y octa-PCDD/PCDF se determinan como
se indica en la siguiente en la ecuación:
![]()
donde:
RRF = Factor de respuesta
relativo calculado.
Qis = Cantidad de estándar
interno adicionado a cada muestra.
As = La respuesta SIM para
los iones nativos de la m/z presentada en la Tabla 17.8.
Ais = La respuesta SIM para el
ión del estándar interno (m/z de la Tabla 17.8).
NOTA: Si
algún factor de dilución se utiliza, debe ser aplicado a estos cálculos.
12.9
Concentración mínima detectable-La concentración mínima detectable para un
isómero individual de tetra, penta, hexa, hepta y octa-PCDDs/PCDFs se determina
con las siguientes ecuaciones:
![]()
donde:
RRF = Factor de respuesta
relativo calculado.
Qis = Cantidad de estándar
interno adicionado a cada muestra.
As = Ruido promedio adyacente
al pico congénere
Ais = La respuesta SIM para el
ión del estándar interno (m/z de la Sección 17.8).
NOTA: Si
algún factor de dilución se utiliza, debe ser aplicado a estos cálculos.
12.10
Factor de respuesta relativo-Este se determina de las siguientes ecuaciones
usando los datos obtenidos de la sección 11.4
del análisis de los estándares de calibración.
![]()
donde:
As = La respuesta SIM para
los iones nativos de la m/z presentada en la Tabla 17.8.
Ais = La respuesta SIM para el
ión del estándar interno (m/z de la Tabla 17.8).
Cs = Concentración del ión
nativo de interés.
Cis = Concentración del
apropiado estándar interno, en ng/µL.
12.11 % de recuperación
de estándares internos-Calcule el % de recuperación, Ris para cada estándar
interno en el extracto de muestra.
![]()
donde:
Ars = Area del ión de cuantificación
del estándar recuperado de 13C12 -1,2,3,4,-TCDD o 13C12
1,2,3,4,7,8-HxCDD.
Qrs = ng de estándar recuperado,
13C12-1,2,3,4,-TCDD o 13C12 1,2,3,4,7,8,-HxCDD
adicionada al extracto.
12.12 Factor de
respuesta para determinar la recuperación: Se calcula de los datos obtenidos de
los análisis de los estándares de calibración.
![]()
donde:
Crs = Concentración
de la recuperación del estándar 13C12 -1,2,3,4-TCDD o 13C12
1,2,3,4,7,8-HCDD.
12.13 Concentración
total de homólogos-Esta se calcula de la concentración de todos los isómeros
con cada serie de homólogos de PCDD’s y PCDF’s usando la siguiente ecuación.
Ecuación 18:
Total de concentración PCDD’s y PCDF’s = Suma
de la concentración individual de isómeros de PCDD’s y PCDF’s
12.14 Reporte de resultados.
Cualquier
desviación del procedimiento descrito debe documentarse en el reporte analítico
de la muestra.
12.14.1 Reporte
Analítico.
Cada
reporte de análisis debe contener tablas de resultados las cuales incluyen lo
siguiente:
12.14.1.1 Identificación
completa de la muestra analizada. La información pertinente del muestreo debe
ser registrada por el laboratorio analítico vía las hojas de campo y cadena de
custodia.
12.14.1.2 Datos de la
recepción de la muestra, fecha y hora de la extracción y limpieza, del análisis
por HRGC/LRMS esta última información debe de aparecer en cada cromatograma de
masas incluido en el reporte.
12.14.1.3 Los datos crudos
del HRGC/LRMS.
12.14.1.4 Cálculos de la
relación de abundancia de los iones moleculares para todas las PCDDs y PCDFs
detectados.
12.14.1.5 Las cantidades de
PCDDs/PCDFs reportadas como nanogramos (ng) por muestra. Los valores del total
de tetra, penta, hexa, hepta y octa-CDDs y CDFs y cada uno de los isómeros
2,3,7,8 sustituidos. Si las PCDDs y PCDFs no se detectan se debe reportar el
valor mínimo detectable.
12.14.1.6 Todos los datos y
documentos analíticos deben ser iguales para las muestras problema y para los
blancos, y demás muestras de control de calidad.
12.14.1.7 La recuperación
de los estándares internos en %.
12.14.1.8 La recuperación
de PCDDs/PCDFs nativos en las muestras adicionadas en %.
12.14.1.9 Los datos de
calibración, incluidos promedios de los factores de respuesta calculados de los
5 puntos de curva, descrito en la sección 10.2.5
incluyendo la desviación estándar y datos que demuestren que estos valores se
han verificado al menos durante un periodo de 12 h de operación o con cada lote
separado de muestras analizadas.
13. Desempeño del Método
13.1 Es la suma de las
concentraciones mínimas detectables de los 17 congéneres expresado en EQT, debe
ser menor a 0,07 ng/m3 en
condiciones estándares de 7% de oxígeno
13.2 Límite Práctico
de Cuantificación: 0,20 ng/m3 EQT
13.3 Rango de Trabajo:
de 0,20 a 5,0 ng/m3 EQT
13.4 Precisión Inicial
del Método: Por determinar
13.5 Exactitud Inicial
del Método: Por determinar
13.6 Precisión
Continua del Método: Por determinar
13.7
Exactitud Continua del Método: Por determinar
13.8
Recuperación de Surrogados: de 60 a 140%
13.9
Recuperación de Estándares internos: de 60 a 120%
13.10 En
caso de interferencias positivas o que no se alcancen los límites mínimos
detectables se deberá utilizar el método de análisis de cromatografía de gases
por alta resolución acoplado a espectrometría de alta resolución.
14.
Prevención de la Contaminación
14.1 La
principal fuente de contaminación es la exposición a los vapores tóxicos de los
disolventes y a la exposición a derrames de los extractos de las muestras por
lo que el laboratorio debe contar con una buena ventilación y el personal debe
trabajar siempre con guantes y equipo de seguridad completo.
14.2
Realizar todas las operaciones de extracción, preparación de estándares y
muestras en la campana de extracción y además usar mascarilla.
15.
Manejo de residuos
15.1 Es
la responsabilidad del laboratorio cumplir con todos los reglamentos federales,
estatales y locales referentes al manejo de residuos, particularmente las
reglas de identificación, almacenamiento y disposición de residuos peligrosos.
15.2 Confinamiento:
El laboratorio debe contar con áreas especiales, que tengan señalamientos
adecuados, para almacenar temporalmente las soluciones y materiales
contaminados con PCDDs/PCDFs.
15.3
Todos los extractos de las muestras deben ser destruidos mediante oxidación
fotolítica con luz UV de onda corta y demostrarse su destrucción mediante
HRGC/ECD.
16. Referencias
16.1 Método
428, “Determination of Polychlorinated Dibenzo-p-Dioxin (PCDD) and
Polychlorinated Dibenzofuran (PCDF) Emissions from Stationary Sources”, State
Of California, Air Resources Board, September 12, 1990.
16.2 Método
23 A, “Determination of Polychlorinated Dibenzo-p-Dioxins and Polychlorinated
Dibenzofurans Emissions from Stationary Sources”, United States Environmental
Protection Agency, August, 1996.
17. Tablas y Figuras
17.1
Analitos monitoreados por este método
|
PCDD’s |
PCDF’s |
|
2,3,7,8-TCDD El
total de las TCDD 1,2,3,7,8-PeCDD Total
de las PeCDD 1,2,3,4,7,8-HxCDD 1,2,3,6,7,8-HxCDD 1,2,3,7,8,9-HxCDD El
total de las HxCDD 1,2,3,4,6,7,8-HpCDD El
total de las HpCDD 1,2,3,4,6,7,8,9-OCDD El
total de las OCDD |
2,3,7,8-TCDF El
total de los TCDF 1,2,3,7,8-PeCDF 2,3,4,7,8-PeCDF El
total de los PeCDF 1,2,3,4,7,8-HxCDF 1,2,3,6,7,8-HxCDF 1,2,3,7,8,9-HxCDF 2,3,4,6,7,8-HxCDF El
total de los HxCDF 1,2,3,4,6,7,8-HpCDF 1,2,3,4,7,8,9-HpCDF El
total de los HxCDF 1,2,3,4,6,7,8,9-OCDF El
total de los OCDF |
17.2 Límites de
detección
|
Congéneres |
Límites de
Detección (pg/muestra) |
|
|
|
|
TCDD/TCDF |
2,000 |
|
PeCDD/PeCDF |
4,000 |
|
HxCDD/HxCDF |
4,000 |
|
HpCDD/HpCDF |
4,000 |
|
OCDD/OCDF |
6,000 |
17.3 Concentración de
PCDD’s y PCDF’s en los estándares de calibración del HRGC/LRMS
|
|
|
Concentración
(pg/µL) |
|
|
|||
|
|
1 |
2 |
3 |
4 |
5 |
||
|
Estándares de
Calibración: |
|
|
|
|
|||
|
2,3,7,8-TCDD |
100 |
200 |
1000 |
2000 |
5000 |
||
|
1,2,3,7,8-PeCDD |
100 |
200 |
1000 |
2000 |
5000 |
||
|
1,2,3,4,7,8-HxCDD |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,6,7,8-HxCDD |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,7,8,9-HxCDD |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,4,6,7,8-HpCDD |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,4,6,7,8,9-OCDD |
500 |
1000 |
5000 |
10000 |
25000 |
||
|
|
|
|
|
|
|
||
|
2,3,7,8-TCDF |
100 |
200 |
1000 |
2000 |
5000 |
||
|
1,2,3,7,8-PeCDF |
100 |
200 |
1000 |
2000 |
5000 |
||
|
2,3,4,7,8-PeCDF |
100 |
200 |
1000 |
2000 |
5000 |
||
|
1,2,3,4,7,8-HxCDF |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,6,7,8-HxCDF |
250 |
500 |
2500 |
5000 |
12500 |
||
|
2,3,4,6,7,8-HxCDF |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,7,8,9-HxCDF |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,4,6,7,8-HpCDF |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,4,7,8,9-HpCDF |
250 |
500 |
2500 |
5000 |
12500 |
||
|
1,2,3,4,6,7,8,9-OCDF |
500 |
1000 |
5000 |
10000 |
25000 |
||
|
|
|
|
|
|
|
||
|
Estándares
Internos: |
|
|
|
|
|
||
|
13C-2,3,7,8-TCDD |
500 |
1000 |
5000 |
10000 |
25000 |
||
|
13C-1,2,3,7,8-PeCDD |
500 |
1000 |
5000 |
10000 |
25000 |
||
|
13C-1,2,3,6,7,8-HxCDD |
500 |
1000 |
5000 |
10000 |
25000 |
||
|
13C-1,2,3,4,6,7,8-HpCDD |
1000 |
2000 |
10000 |
20000 |
50000 |
||
|
13C-1,2,3,4,6,7,8,9-OCDD |
1000 |
2000 |
10000 |
20000 |
50000 |
||
|
|
|
|
|
|
|
||
|
13C-2,3,7,8-TCDF |
500 |
1000 |
5000 |
10000 |
25000 |
||
|
|
|
|
|
|
|
||
|
Estándares
surrogados: |
|
|
|
|
|||
|
37Cl-2,3,7,8-TCDD |
100 |
200 |
1000 |
2000 |
5000 |
||
|
13C-1,2,3,7,8,9-HxCDD |
250 |
500 |
2500 |
5000 |
12500 |
||
|
13C-1,2,3,4,6,7,8-HpCDF |
250 |
500 |
2500 |
5000 |
12500 |
||
|
|
|
|
|
|
|
||
|
Estándares de
Recuperación: |
|
|
|
|
|||
|
13C12-1,2,3,4-TCDD |
500 |
500 |
500 |
500 |
500 |
||
|
13C12-1,2,3,4,7,8-HxCDD |
1000 |
1000 |
1000 |
1000 |
1000 |
||
17.4 Concentración de
estándares internos en el extracto de la muestra
|
ESTANDAR
INTERNO |
pg/µL |
|
|
|
|
13C-TCDD |
500 |
|
13C-PeCDD |
500 |
|
13C-HxCDD |
500 |
|
13C-HpCDD |
1000 |
|
13C-OCDD |
1000 |
|
13C-TCDF |
500 |
17.5 COMPUESTOS
CONTENIDOS EN LA MEZCLA DE EVALUACION DE LA COLUMNA.
|
TCDD |
1,3,6,8; |
1,2,8,9; |
2,3,7,8; |
|
PeCDD |
1,2,4,6,8; |
1,2,3,8,9 |
|
|
HxCDD |
1,2,3,4,6,9; |
1,2,3,4,6,7 |
|
|
HpCDD |
1,2,3,4,6,7,8; |
1,2,3,4,6,7,9 |
|
|
OCDD |
1,2,3,4,6,7,8,9 |
|
|
|
TCDF |
1,3,6,8; |
1,2,8,9 |
|
|
PeCDF |
1,3,4,6,8; |
1,2,3,8,9 |
|
|
HxCDF |
1,2,3,4,6,8; |
1,2,3,4,7,8,9 |
|
|
HpCDF |
1,2,3,4,6,7,8; |
1,2,3,4,7,8,9 |
|
|
OCDF |
1,2,3,4,6,7,8,9. |
|
|
Cualquier
mezcla de PCDD/PCDF que se demuestre contenga estos componentes puede
sustituirse.
17.6 Relación de
estándares interno para la cuantificación y el cálculo de RRF de la
determinación de dioxinas y furanos.
|
PCDF/PCDD Nativas |
ISTD’s de cuantificación |
Estándar de Calibración |
|
TCDD |
13C-2,3,7,8-TCDD |
2,3,7,8-TCDD |
|
PeCDD |
13C-1,2,3,7,8-PCDD |
1,2,3,7,8-PeCDD |
|
HxCDD |
13C-1,2,3,6,7,8-HxCDD |
2,3,7,8-,X,Y-HxCDD |
|
HpCDD |
13C-1,2,3,4,6,7,8-HpCDD |
1,2,3,4,6,7,8-HpCDD |
|
OCDD |
13C-OCDD |
OCDD |
|
TCDF |
13C-2,3,7,8-TCDF |
2,3,7,8-TCDF |
|
PeCDF |
13C-1,2,3,7,8-PeCDD |
2,3,7,8,X-PeCDF |
|
HxCDF |
13C-1,2,3,6,7,8-HxCDD |
2,3,7,8-X,Y
-HxCDF |
|
HpCDF
|
13C-1,2,3,4,6,7,8-HpCDD |
2,3,7,8-X,Y,Z-HpCDF |
|
OCDF |
13C-OCDD |
OCDF |
17.7
Condiciones cromatográficas recomendadas.
|
|
SP2331
60 m |
DB-5 60
m |
|
Velocidad lineal de flujo |
- |
30 cm/seg |
|
Temperatura inicial |
170ºC |
190ºC |
|
Tiempo inicial |
1 min |
1 min |
|
Tiempo de Splitless |
0.6
min |
0.6
min |
|
Rampa de temperatura/tiempo |
10ºC/min |
8ºC/min |
|
Temperatura final |
250ºC |
300ºC |
|
Tiempo final |
15 min |
7 min |
|
Flujo del split |
30 mL/min |
30 mL/min |
|
Flujo de la purga de septa |
5 mL/min |
5 mL/min |
|
Presión de la cabeza de la columna |
28 psi |
15 psi |
17.8
Iones específicos para la selección del monitoreo de iones para PCDD’s y PCDF’s
y relaciones isotópicas.
|
Compuestos |
Masas a monitorear |
Relación
de isótopos teóricos [M]+:[M+2]+ o [M+2]+:[M+4]+ |
|
|
PCDDs: |
|
|
|
|
TCDD |
319.89 |
321.89 |
0.77 |
|
13C12-TCDD |
331.9368 |
333.93 |
0.77 |
|
|
|
|
|
|
PeCDD |
355.85 |
357.85 |
1.54 |
|
13C12-PeCDD |
367.89 |
369.89 |
1.54 |
|
|
|
|
|
|
HxCDD |
387.81 |
389.81 |
|
|
|
389.81 |
391.81 |
|
|
13C12-HxCDD |
391.85 |
393.85 |
1.23 |
|
|
|
|
|
|
HpCDD |
423.77 |
425.77 |
1.03 |
|
13C12-HpCDD |
435.81 |
437.81 |
1.03 |
|
|
|
|
|
|
OCDD |
457.73 |
459.73 |
0.88 |
|
13C12-OCDD |
469.77 |
471.77 |
0.88 |
|
|
|
|
|
|
PCDFs: |
|
|
|
|
TCDF |
303.90 |
305.89 |
0.77 |
|
13C-12-TCDF |
315.94 |
317.93 |
0.77 |
|
|
|
|
|
|
PeCDF |
339.89 |
341.85 |
1.54 |
|
|
|
|
|
|
HxCDF |
371.82 |
373.82 |
|
|
|
373.82 |
375.81 |
1.23 |
|
|
|
|
|
|
HpCDF |
407.78 |
409.77 |
1.03 |
|
13C12-HpCDF |
419.82 |
421.81 |
1.03 |
|
|
|
|
|
|
OCDF |
441.74 |
443.73 |
0.88 |
|
|
|
|
|
NOTA: Los iones m/z
374, 376, 378 (HxCDE), 410 (HpCDE), 446 (OCDE), 480 (NCDE) y 514 (DCDE) deben
incluirse en el monitoreo (interferencias más comunes).
17.9 Estándares de
calibración y estándares internos para el cálculo de RFF y la cuantificación de
PCDDs y PCDFs en una muestra de gases de chimenea.
|
PCDD/PCDF |
Estándar interno para calcular los FRR y
cuantificar los compuestos nativos |
Estándar de Calibración |
|
PCDDs/PCDFs
nativas: |
|
|
|
TCDD |
13C-2,3,7,8-TCDD |
2,3,7,8-TCDD |
|
PeCDD |
13C-1,2,3,7,8-PeCDD |
1,2,3,7,8-PeCDD |
|
HxCDD |
13C-1,2,3,6,7,8-HxCDD |
2,3,7,8,X,Y-HxCDD |
|
HpCDD |
13C-1,2,3,4,6,7,8-HpCDD |
1,2,3,4,6,7,8-HpCDD |
|
OCDD |
13C-OCDD |
OCDD |
|
TCDF |
13C-2,3,7,8-TCDF |
2,3,7,8-TCDF |
|
PeCDF |
13C-1,2,3,7,8-PeCDD |
2,3,7,8,X-PeCDF |
|
HxCD |
13C-1,2,3,6,7,8-HxCDD |
2,3,7,8,X,Y-HxCDF |
|
HpCDF |
13C-1,2,3,4,6,7,8-HpCDD |
2,3,7,8,X,Y,Z-HpCDF |
|
OCDF |
13C-OCDD |
OCDF |
|
|
|
|
|
Estándares
Surrogados: |
|
|
|
37Cl-1,2,3,4-TCDD |
13C-2,3,7,8-TCDD |
|
|
13C-1,2,3,7,8,9-HxCDD |
13C-1,2,3,6,7,8-HxCDD |
|
|
13C-1,2,3,4,6,7,8-HpCDF |
13C-1,2,3,4,6,7,8-HpCDD |
|
|
|
|
|
|
Estándares
de Recuperación: |
|
|
|
13C12-1,2,3,4-TCDD |
13C-2,3,7,8-TCDD |
|
|
13C12-1,2,3,4,7,8-HxCDD |
13C-1,2,3,6,7,8-HxCDD |
|
|
|
|
|
17.10 Congéneres de
PCDDs/PCDFs isotópicamente marcados para ser utilizados en el muestreo y
análisis de PCDDs/PCDFs.
|
Estándares
Surrogados1 |
Estándares
Internos2 |
Estándares
de Recuperación3 |
|
37Cl4-2,3,7,8-TCDD |
13C12-2,3,7,8-TCDD |
13C12-1,2,3,4-TCDD |
|
13C12-1,2,3,7,8,9-HxCDD |
13C12-1,2,3,7,8-PeCDD |
13C12-1,2,3,4,7,8-HxCDD |
|
13C12-1,2,3,4,6,7,8-HpCDF |
13C12-1,2,3,6,7,8-HxCDD |
|
|
|
13C12-1,2,3,4,6,7,8-HpCDD |
|
|
|
13C12-OCDD |
|
|
|
13C12-2,3,7,8-TCDF |
|
1
Adicionado a la resina XAD-2 antes del muestreo.
2
Estándares internos adicionados antes de la extracción.
3
Estándar interno adicionado justo antes de la inyección al HRGC/LRMS.
FIGURA 1
ESQUEMA DE
RECUPERACION DE MUESTRAS DEL TREN DE MUESTREO